ГОСТ ISO 734-1-2016 Жмыхи и шроты. Определение содержания сырого жира. Часть 1. Метод экстракции гексаном (или легким петролейным эфиром). Эфир петролейный гост
ГОСТ 25163-82
ГОСТ 25163-82(СТ СЭВ 2342-80)
Группа Л29
Метод определения свободных полиэтиленгликолейи активного вещества в неионогенных ПАВ
Срок действия с 01.07.1982до 01.07.1989*________________* Ограничение срока действия снятопо протоколу Межгосударственного Советапо стандартизации, метрологии и сертификации.(ИУС N 2 1993 г.).Примечание "КОДЕКС"
РАЗРАБОТАН Министерством нефтеперерабатывающей и нефтехимической промышленности СССРИСПОЛНИТЕЛИ
Г.А.Тембер, Л.В.Макарова, В.Н.Иванов, Т.А.МартыноваВНЕСЕН Министерством нефтеперерабатывающей и нефтехимической промышленности СССРЧлен Коллегии П.А.ВерновУТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Постановлением Государственного комитета СССР по стандартам от 9 марта 1982 г. N 967Настоящий стандарт распространяется на неионогенные поверхностно-активные вещества типа алкил- и алкилфенолполиоксиэтилатов, соответствующих формулам 1 и 2, среднее содержание оксиэтиленовых групп в которых от 2 до 10, и устанавливает метод определения массовых долей свободных полиэтиленгликолей и активного вещества.
(1)
, (2)
где - алкильная группа с прямой или разветвленной цепью, содержащая от 10 до 18 атомов углерода; - алкильная группа с прямой или разветвленной цепью, в основном нонил или третичный октил; - среднее количество оксиэтиленовых групп в одной молекуле.Сущность метода заключается в экстрагировании раствора пробы в этилацетате водным раствором хлористого натрия, при котором полиэтиленгликоли переходят в водную фазу, а неионогенное поверхностно-активное вещество - в органическую фазу, и определении массовой доли неионогенного поверхностно-активного вещества выпариванием этилацетатного экстракта, а массовой доли полиэтиленгликолей - экстракцией водного экстракта хлороформом и выпариванием хлороформного экстракта.Стандарт полностью соответствует СТ СЭВ 2342-80.
1. АППАРАТУРА И РЕАКТИВЫ
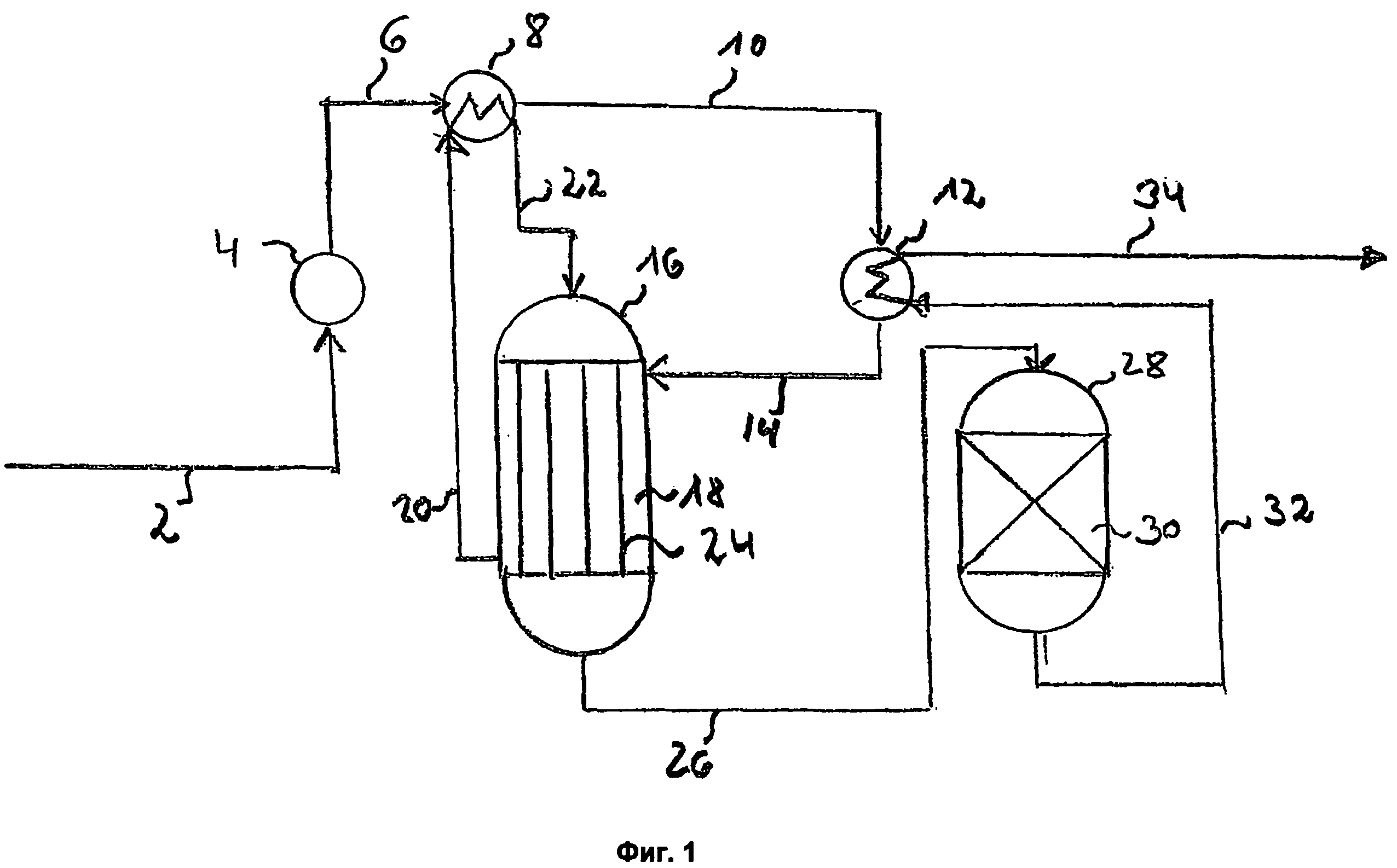
Воронка делительная по ГОСТ 8613-75, вместимостью 250 и 500 см или воронка делительная с рубашкой для термостатирования (чертеж).
Чертеж
Делительная воронка с рубашкой для термостатирования
Колбы КПКШ-250-29/32 ТС, КПКШ-500-29/32 ТС ГОСТ 10394-72.Цилиндр по ГОСТ 1770-74, вместимостью 100 см.Холодильник стеклянный лабораторный по ГОСТ 9499-70.Стакан химический по ГОСТ 10394-72, вместимостью 100 см.Фильтр беззольный "синяя лента", диаметром 9 см.Этилацетат по ГОСТ 8981-78.Хлороформ по ГОСТ 20015-74 или фармакопейный.Ацетон по ГОСТ 2603-79.Эфир петролейный по ГОСТ 11992-66, с пределами выкипания 40-60 °С.Вода дистиллированная по ГОСТ 6709-72.Натрий хлористый, раствор готовят следующим образом: 300 г хлористого натрия, взвешенного с погрешностью не более 0,1 г, растворяют в мерной колбе вместимостью 1000 см в 800 см дистиллированной воды и доводят объем до метки.
2. ПОДГОТОВКА К АНАЛИЗУ
2.1. Все операции растворения и экстракции пробы проводят при температуре (35±1) °С. Используемые аппаратура, реактивы и растворы должны иметь температуру (35±1) °С.
2.2. Экстракцию полиэтиленгликолей из водного раствора хлористого натрия хлороформом допускается проводить при температуре окружающей среды (20±5) °С.
2.3. Для достижения необходимой температуры анализа работают в закрытой камере с температурой (35±1) °С или применяют делительные воронки с рубашкой для термостатирования при (35±1) °С.
3. ПРОВЕДЕНИЕ АНАЛИЗА
3.1. (5±0,05) г пробы взвешивают с погрешностью не более 0,01 г, растворяют в стакане в таком объеме этилацетата, чтобы общий объем раствора пробы составил 75 см, и переносят в делительную воронку А вместимостью 250 см. В воронку прибавляют 50 см водного раствора хлористого натрия, встряхивают в течение 1 мин и выдерживают не менее 30 мин до четкого разделения фаз. Водный экстракт (нижний слой) сливают в делительную воронку Б вместимостью 250 см. Экстракцию повторяют три раза, прибавляя каждый раз по 50 см водного раствора хлористого натрия, и водный экстракт собирают в воронку Б. Затем в воронку Б прибавляют 25 см этилацетата и экстрагируют, как указано выше, собирая водный экстракт (нижний слой) в воронку В вместимостью 250 см.Затем в воронку В добавляют 25 см этилацетата и экстрагируют, как указано выше, собирая водный экстракт (нижний слой) в делительную воронку Г вместимостью 500 см, а органическую фазу (верхний слой) переносят в воронку Б, в которой также находится органическая фаза. Затем в воронку Б добавляют 25 см водного раствора хлористого натрия и экстрагируют, как указано выше.После этого воронку В промывают водным экстрактом, полученным в воронке Б, и переносят его в воронку Г. Воронку В промывают 10 см свежего водного раствора хлористого натрия и промывную жидкость также сливают в воронку Г.Таким образом, в воронку Г собирают весь водный экстракт пробы, содержащий полиэтиленгликоль, а в воронки А и Б собирают органический экстракт, содержащий активное веще
ство.
3.2. Содержимое воронки А переносят в перегонную колбу 1 вместимостью 250 см с пришлифованной пробкой и отгоняют этилацетат. Содержимым воронки Б промывают воронку А и переносят его в перегонную колбу 1.Воронки А и Б последовательно, начиная с воронки Б, два раза промывают порциями этилацетата по 25 см и переносят промывную жидкость в перегонную колбу 1. Из содержимого перегонной колбы отгоняют этилацетат, после чего к сухому остатку добавляют 75 см этилацетата, подогретого до 45 °С, растворяют сухой остаток и сразу фильтруют через фильтровальную бумагу в перегонную колбу 2 вместимостью 250 см, предварительно взвешенную с погрешностью не более 0,001 г.Перегонную колбу 1 и фильтр 1 промывают шесть раз порциями этилацетата по 10 см, нагретого до 35 °С. Фильтраты собирают вместе в перегонную колбу 2.Этилацетат из перегонной колбы 2 отгоняют на кипящей водяной бане, а остаток выпаривают досуха. К сухому остатку прибавляют 10 см ацетона и снова выпаривают досуха. Выпаривание с добавлением 10 см ацетона повторяют еще раз, а затем к сухому остатку добавляют 10 см петролейного эфира и выпаривают досуха. Перегонную колбу 2 закрывают и ставят в сушильный шкаф, нагретый до (100±5) °С на 10 мин, продувают струей воздуха и взвешивают с погрешностью не более 0,001 г.Операции, начиная с прибавления 10 см ацетона, повторяют до тех пор, пока разница между двумя последовательными взвешиваниями будет равна или будет менее 0,005
г.
3.3. В делительную воронку Г, содержащую водную фазу, добавляют 100 см хлороформа, встряхивают в течение 1 мин и выдерживают не менее 15 мин. Затем переносят хлороформный слой в перегонную колбу вместимостью 500 см. Экстракцию повторяют два раза, используя каждый раз 100 см хлороформа.Из собранных экстрактов отгоняют на водяной бане хлороформ и затем выпаривают досуха. Сухой остаток растворяют в 50 смхлороформа. Полученный раствор фильтруют через фильтровальную бумагу в предварительно взвешенную перегонную колбу 3 вместимостью 250 см. Перегонную колбу 1 и фильтр промывают шесть раз порциями по 10 см хлороформа, нагретого до 35 °С. Из собранного фильтрата отгоняют хлороформ (хлороформ можно использовать повторно для экстракции из водного раствора после стабилизации 2 см этилового спирта на 100 см хлороформа), сухой остаток досушивают, дважды добавляя по 10 см ацетона и 10 см петролейного эфира, и взвешивают.Операции, начиная с прибавления 10 см ацетона, повторяют до тех пор, пока разница между двумя последовательными взвешиваниями будет равна или будет менее 0,005 г.
4. ОБРАБОТКА РЕЗУЛЬТАТОВ
4.1. Массовую долю свободных полиэтиленгликолей () в процентах вычисляют по формуле
,
где - масса навески пробы, г; - масса сухого остатка, полученная по п.3.3, г.
4.2. Массовую долю активного вещества () в процентах вычисляют по формуле
,
где - масса навески пробы, г; - масса сухого остатка, полученная по п.3.2, г.
4.3. За результат анализа принимают среднее арифметическое двух параллельных определений, допускаемое расхождение между которыми не должно превышать 0,3% для полиэтиленгликолей и 1% для активного вещества.Текст документа сверен по:официальное изданиеМ.: Издательство стандартов, 1982
docs.cntd.ru
ГОСТ ISO 734-1-2016 Жмыхи и шроты. Определение содержания сырого жира. Часть 1. Метод экстракции гексаном (или легким петролейным эфиром), ГОСТ от 30 августа 2016 года №ISO 734-1-2016
ГОСТ ISO 734-1-2016
МКС 67.200
Дата введения 2018-01-01
Цели, основные принципы и основной порядок проведения работ по межгосударственной стандартизации установлены ГОСТ 1.0-2015 "Межгосударственная система стандартизации. Основные положения" и ГОСТ 1.2-2015 "Межгосударственная система стандартизации. Стандарты межгосударственные, правила и рекомендации по межгосударственной стандартизации. Правила разработки, принятия, обновления и отмены"Сведения о стандарте
1 ПОДГОТОВЛЕН Федеральным государственным бюджетным научным учреждением "Всероссийский научно-исследовательский институт жиров" (ВНИИЖиров) на основе официального перевода на русский язык англоязычной версии международного стандарта, указанного в пункте 4, который выполнен ФГУП "СТАНДАРТИНФОРМ"
2 ВНЕСЕН Федеральным агентством по техническому регулированию и метрологии
3 ПРИНЯТ Межгосударственным советом по стандартизации, метрологии и сертификации (протокол от 28 июня 2016 г. N 49)За принятие проголосовали:
| Краткое наименование страны по МК (ИСО 3166) 004-97 | Код страны поМК (ИСО 3166) 004-97 | Сокращенное наименование национального органа по стандартизации |
| Армения | AM | Минэкономики Республики Армения |
| Киргизия | KG | Кыргызстандарт |
| Россия | RU | Росстандарт |
4 Приказом Федерального агентства по техническому регулированию и метрологии от 30 августа 2016 г. N 962-ст межгосударственный стандарт ГОСТ ISO 734-1-2016 введен в действие в качестве национального стандарта Российской Федерации с 1 января 2018 года.
5 Настоящий стандарт идентичен международному стандарту ISO 734-1:2006* "Жмыхи и шроты. Определение содержания масла. Часть 1. Метод экстракции гексаном (или легким петролейным эфиром)" (Oilseed meals - Determination of oil content - Part 1: Extraction method 1 with hexane (or light petroleum)", IDT).________________* Доступ к международным и зарубежным документам, упомянутым здесь и далее по тексту, можно получить, перейдя по ссылке на сайт http://shop.cntd.ru. - Примечание изготовителя базы данных.Международный стандарт разработан Техническим комитетом по стандартизации ISO/TC 34 "Пищевые продукты" Международной организации по стандартизации (ISO).Наименование настоящего стандарта изменено относительно наименования указанного международного стандарта для приведения в соответствие с общепринятой терминологией и ГОСТ 1.5 (подраздел 3.6).При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им межгосударственные стандарты, сведения о которых приведены в дополнительном приложении ДА.
6 ВВЕДЕН ВПЕРВЫЕИнформация об изменениях к настоящему стандарту публикуется в ежегодном информационном указателе "Национальные стандарты", а текст изменений и поправок - в ежемесячном информационном указателе "Национальные стандарты". В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ежемесячном информационном указателе "Национальные стандарты". Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования - на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.gost.ru)
1 Область применения
Настоящий стандарт устанавливает метод определения веществ, экстрагируемых гексаном (или легким петролейным эфиром), называемых "содержанием сырого жира" в жмыхах и шротах (за исключением смешанных продуктов), полученных при извлечении масла из масличных семян прессованием или экстракцией растворителем.
2 Нормативные ссылки
Следующие ссылочные нормативные документы являются обязательными при применении данного документа*. Для датированных ссылок применяется только цитированное издание документа. Для недатированных ссылок необходимо использовать самое последнее издание нормативного ссылочного документа (включая любые изменения).________________* Таблицу соответствия национальных стандартов международным см. по ссылке. - Примечание изготовителя базы данных.ISO 771 Oilseed residues - Determination of moisture and volatile matter content (Жмыхи и шроты. Определение содержания влаги и летучих веществ)ISO 5502, Oilseed residues - Preparation of test samples (Жмыхи и шроты. Подготовка образца)
3 Термины и определения
В настоящем стандарте применен термин с соответствующим определением.
3.1 содержание сырого жира (oil content): Все вещества, экстрагируемые в условиях, установленных настоящим стандартом, и выраженные в виде массовой доли, в процентах.Примечание - Содержание сырого жира также может быть выражено в пересчете на сухое вещество.
4 Сущность метода
Пробу для анализа экстрагируют в специальном аппарате техническим гексаном или, при его отсутствии, легким петролейным эфиром. Отгоняют растворитель, и остаток взвешивают.
5 Реактивы
Используют реактивы только известной аналитической степени чистоты, если не указано иное.
5.1 Технический гексан, н-гексан или легкий петролейный эфир, состоящий, в основном, из углеводородов с шестью атомами углерода.Менее 5% растворителя должно перегоняться при температуре ниже 50°С, более 95% - при температуре от 50°С до 70°С.Для любого из этих растворителей остаток при полном выпаривании не должен превышать 2 мг на 100 см.
6 Аппаратура
Обычное лабораторное оборудование и, в частности, следующее.
6.1 Механический измельчитель, легко очищаемый, измельчающий материал без нагрева и ощутимого изменения содержания влаги и летучих веществ и содержания масла, обеспечивающий получение частиц, полностью проходящих через сито с размером отверстий 1 мм.
6.2 Механический микроизмельчитель типа Dangoumau*, обеспечивающий степень измельчения материала менее 160 мкм, за исключением "шелухи", частицы которой могут достигать 400 мкм._______________* Механический микроизмельчитель Dangoumau является примером подходящего оборудования, имеющегося в продаже. Эта информация приводится для удобства пользователей настоящего стандарта и не связана с поддержкой этого оборудования.При отсутствии микроизмельчителя дополнительное измельчение пробы (см. 9.4.3) может быть заменено растиранием с помощью пестика и ступки в присутствии приблизительно 10 г песка, промытого соляной кислотой и затем прокаленного.Однако растирание в ступке не может применяться в случае многократных анализов, потому что усталость оператора мешает эффективному измельчению многочисленных проб, а экстрагирование масла из крупноизмельченной пробы никогда не может быть полным.
6.3 Экстракционный патрон и хлопковая вата или фильтровальная бумага, не содержащие веществ, растворимых в гексане или легком петролейном эфире.
6.4 Экстракционный аппарат, снабженный колбой вместимостью от 200 см до 250 см.Примечание - Пригодны прямоточные экстракторы, например экстракторы Butt, Smalley, Twisselmann и Bolton-Williams*. Допускается использование других экстракторов при условии, что результаты испытания на стандартном материале с известным содержанием масла подтверждают пригодность аппарата._______________* Прямоточные экстракторы Butt, Smalley, Twisselmann или Bolton-Williams являются примерами подходящего оборудования, имеющегося в продаже. Эта информация дается для удобства пользователей настоящего стандарта и не связана с поддержкой этого оборудования.
6.5 Электрическая нагревательная баня (например, песчаная или водяная) или нагревательная плитка.
6.6 Электрический термостат, обеспечивающий вентилирование или получение пониженного давления, позволяющий поддерживать температуру 103°С±2°С.
6.7 Эксикатор, содержащий эффективный осушитель.
docs.cntd.ru
Петролейный эфир определение воды - Справочник химика 21
Известны разнообразные другие цветные реакции с пирогалловой кислотой, трихлоруксусной кислотой, сахарозой, дихлоргидрином глицерина (зеленая окраска с Хмакс 625 нм), ароматическими альдегидами и др. они предложены для качественного и количественного определения витамина В в витаминных концентратах и пищевых продуктах [68]. Описан метод количественного определения витаминов Ог н Од хроматографией на бумаге с применением в качестве подвижной фазы петролейного эфира, насыщенного водой [69]. [c.104] Для определения общего содержания органических веществ навеску образца (10 г) перемешивают с 50 мл дистиллированной воды, подкисленной 10%-й азотной кислотой, и экстрагируют из этой смеси органические вещества петролейным эфиром в тех же условиях, что и при извлечении нафтеновых кислот. После промывки экстракта органических веществ в петролейном эфире дистиллированной водой и отгонки растворителя органические вещества высушивают до постоянной массы. [c.30]Сборка приборов. Перед определением вязкости вискозиметр должен быть тщательно промыт петролейным эфиром или легким бензином, хромовой смесью, дистиллированной водой и просушен чистым воздухом. [c.231]
Устанавливают прибор так, чтобы зеркало было освещено дневным рассеянным (или искусственным) светом, и в случае необходимости пускают ток воды для обеспечения нужной температуры определения. После этого раскрывают призмы, промывают их петролейным эфиром, а затем протирают сухой и мягкой замшей или льняной тряпочкой. [c.81]
Выяснение возможных путей циркуляции ароматических углеводородов в различных объектах окружающей среды осуществлялось в экспериментальных моделях. Для этих целей специально были разработаны высокочувствительные газохроматографические методы раздельного определения исследуемых веществ в почве, растениях, воде и воздухе. Предварительная подготовка проб заключалась в извлечении петролейным эфиром ароматических углеводородов из почвы, растений и воды с последующей очисткой и концентрированием. [c.85]
Определение растворимости вещества проводят с несколькими растворителями, причем соблюдают следующую очередность вода, эфир, 5% -ный раствор едкого натра, 5%-ный раствор бикарбоната иатрия, 5%-ный раствор соляной кислоты, концентрированная серная кислота. Далее в качестве растворителя испытывают спирт, бензол, ледяную уксусную кислоту, петролейный эфир (чтобы найти растворитель для перекристаллизации и разделения смесей). [c.295]
Перед определением вязкости мазута его предварительно обезвоживают и освобождают от механических примесей. Внутренний сосуд вискозиметра тщательно промывают серным или петролейным эфиром, затем спиртом и под конец дистиллированной водой. Прибор устанавливают таким образом, чтобы все 3 штифта во (Внутреннем измерительном сосуде находились в горизонтальной плоскости, для чего регулируют установочные винты треножника до тех пор, пока все три острия указателей уровня будут едва заметны над поверхностью воды, налитой во внутренний сосуд в соответствующем объеме. Этот объем регулируется отнятием или добавлением жидкости при помощи пипетки. Установленный таким образом треножник не следует сдвигать с места. Нагревательную баню вискозиметра необходимо каждый раз устанавливать на треножнике всегда в одном и том же положении. [c.243]
Отфильтрованный раствор был сконцентрирован в вакууме в присутствии ацетальдегида и дал продукт с выходом 18 г. На основании рентгенограммы можно предположить, что это был кристаллический нитролигнин, содержавший 53,8% углерода, 5,5% водорода, 3,7% азота и 8,2% метоксилов. Молекулярный вес нитролигнина, равный 1140, был определен криоскопическим методом в диоксане, а также на основе трех метоксильных групп, и содержания азота. Продукт не растворялся в воде, эфире, бензоле и петролейном эфире. Он растворялся в разбавленных калийной и натриевой щелочах, в уксусной кислоте, диоксане, бензальдегиде, в этиловом спирте и уксусном эфире. Продукт разлагался при 175° инфракрасный спектр был аналогичен спектру других лигнинных препаратов. [c.350]
Было исследовано также влияние растворителя на колориметрическое определение сложных эфиров. Поскольку нередко необходимы или желательны другие растворители, помимо этанола были исследованы некоторые из них (рис. 3.3). Растворы реактива, содержащего Ре +, и пробы сложного эфира в изопропаноле оказались во всех отношениях сравнимы с этанольными. Можно растворять пробу в бензоле, оставляя все прочие реактивы без изменения. Диоксан при соответствующей очистке мог бы, вероятно, оказаться также подходящим растворителем. Успешно применяли смеси хлористого метилена с этанолом и петролейный эфир. Томпсон [14] пользовался диэтиловым эфиром после весьма тщательной его очистки. По-видимому, при соответствующей очистке можно применять в качестве растворителей и другие простые эфиры и спирты. Можно анализировать и водные растворы сложных эфиров, однако в этом случае наблюдается некоторое ослабление интенсивности окраски, вероятно, из-за конкурирующих взаимодействий воды и гидроксамовой кислоты с ионами Ре +. Для построения калибровочных кривых всегда необходимо пользоваться тем же растворителем, что и при анализе пробы. [c.146]
Определение количества никотина в табаке на фабриках никотина производится обычно по методу Тота (ТоШ). Результаты не всегда совершенно точны, но достаточны для целей производства. Растирают 6 г порошка табака с 20 сл 20%-ного едкого натра в ступке, которую затем покрывают стеклянной пластинкой, и дают стоять % часа. По. истечении этого срока быстро прибавляют столько порошка гипса, чтобы получить сухой порошок. Его переносят в склянку и заливают смесью из 50 сл эфира и 50 сл петролейного эфира. Взбалтывают в течение часа по крайней мере 50 раз, отфильтровывают 20 сл% прибавляют к ним 50 сл воды и 5 сл1 0,Ш серной кислоты., Избыток кислоты оттитровывают обратно 0,Ш щелочью, индикатором служит иод-эозин. 1 С/И 0,1/У серной кислоты соответствует 0,0162 г никотина. [c.348]
Определение водного числа (константы) прибора. Внутренний сосуд прибора тщательно промывают серным или петролейным эфиром, затем спиртом и под конец — дистиллированной водой. При определении следует пользоваться не бывшей в употреблении деревянной палочкой. Если таковой не окажется, то палочку, бывшую в употреблении, следует тщательно промыть эфиром, спиртом и водой. Внутренней поверхности прибора касаться пальцами, бумагой или тряпочкой не следует только в крайнем случае можно протирать ее замшей. Затем во внутренний сосуд, немного выше остриев крючков, наливают дистиллированную воду с температурой +20° С. При помощи водяной бани температуру воды во внутреннем сосуде в течение 10 мин поддерживают на уровне 20° С, потом слегка приподнимают палочку и выпускают из сосуда немного воды при этом вся сточная трубка также заполняется водой. [c.273]
Отгонку воды с толуолом с последующим определением ее количества в дистилляте по реакции с хлористым ацетилом (см. гл. 2) применяли для определения влаги в жирах, восках и маслах [185]. Для содержащих жиры материалов часто удается провести определение воды и жира в одной и той же пробе. Воду отделяют отгонкой с петролейным эфиром, ксилолом или галогенсодержащим углеводородом. Остаток после перегонки содержит раствор жира в применяемом растворителе и нерастворимые компоненты, не содержащие жира. Количество этих компонентов после их выделения, промывки и удаления следов растворителя нагреванием может быть найдено гравиметрическим методом [93, 165, 237]. [c.271]
Разработана методика определения азота после перевода его в пиролитической печи в аммиак в токе увлажненного водорода при 800 °С над гранулированным никелем. Продукты пиролиза пропускают при 430 °С через скруббер с СаО, в котором происходит поглощение кислых га.эов. После скруббера газы пропускают через раствор электролита кулонометрической ячейки. Поглощенный в ячейке аммиак титруют электрогенерированными ионами Н+ [394]. Реакция превращения азота в аммиак (метод Кьельдаля) с последующим его титрованием электрогенерированными Н+ или ОН- используют для определения N2 в сталях и металлическом титане [389], петролейном эфире [390], воде [396] и различных природных соединениях [396, 397]. В работе [393] растворы аммонийных солей, образующихся после разложения пробы, пропускают через колонку, заполненную катионообменной смолой в Н-форме, и кулонометрически титруют выделяющиеся кислоты электрогенерированными ОН -ионами. Конечную точку титрования устанавливают по переходу окраски метилового красного или фенолфталеина. [c.71]
Согласно ГОСТ 1520-42 это определение проводится следующим образом. Пробу нефтепродукта, подогретую до 40—50°С, перемешивают в течение 5 мин. в склянке, отбирают в делительную норонку 3 мл от этой пробы, смешивают 50 мл петролейного эфира или бензина и прибавляют 25 мл дистиллированной воды. Содержимое воронки тщательно перемешивают 5 мин., дают отстояться и спускают нижний слой в стакан. Затем стеклянной палочкой наносят иа индикаторную бумагу (фильтровальная бумага, обработанная 10—15 %-ным водным раствором солянокислого или уксуснокислого анилина) несколько капель водной вытяжки и наблюддют изменение окраски бумаги. Красная окраска указывает на присутствие фурфурола в продукте, отсутствие окраски — на отсутствие его. [c.683]
Определение проводится следующим образом. Навеску содержащего фурфурол нефтепродукта разбавляют 50—100 мл петролейного эфира (выкипающего до 60°), прибавляют 50 мл дистиллированнной воды и взбалтывают в течение 5 мии. [c.683]
В справочнике Нефти СССР (тт. 1—4) приведены данные о содержании парафина в нефтях, полученные по методике ВНИИ НП. Согласно этой методике, определению также предшествует доасфальтизация, осуществляемая посредством обработки пефти (без перегонки) пропаном или петролейным эфиром. Навеску деасфальтированной нефти (2—3 г) растворяют в смеси из 65% (об.) бензола и 35% (растворении нефть подогревают на водяной бане, после чего постепенно охлаждают раствор в охлаждающей смеси (до —21 °С) с последующим холодным фильтрованием для отделения выпавших парафинов. Затем парафины извлекают из фильтра, обогревая кожух воронки водой остатки ппрафинов смывают горячим бензолом. [c.59]
Определение растворимости. Растворимость вещества в различных растворителях помогает сделать заключение о наличии в веществе тех или иных функциональных групп. Кроме того, определение растворимости позволяет подобрать подходящий растворитель для перекристаллизации вещества ( подобное растворяется в подобном ). Растворимость целесообразно исследовать в следующих растворителях вода 5%-ные растворы едкого натра, гидрокарбоиата натрия, соляной кислоты концентрированная серная кислота этиловый спирт бензол петролейный эфир уксусная кислота. В пробирку вносят каплю жидкого или 0,01 г твердого соединения и по каплям 0,2 мл растворителя. После каждой прибавленной порции растворителя смесь взбалтывают. Если соединение полностью растворимо, то его регистрируют как растворимое. Если вещество плохо растворяется или не растворяется при комнатной температуре, нагревают до кипения. В случае плохой растворимости в неорганических растворителях нерастворившееся вещество отделяют, а раствор нейтрализуют и наблюдают, не выделяется ли из него исходное соединение. Помутнение нейтрализуемого фильтрата указывает на свойства вещества кислые — если растворителем была щелочь или сода основные — кислый растворитель. При внесении вещества в раствор гидрокарбоната нужно обратить внимание, не выделяется ли двуокись углерода. [c.122]
Методика определения. Приготовление ацетилирован-ной гидрофобной бумаги. Лист фильтровальной бумаги № 2 размером 10 X 35 см для удаления катионов тяжелых металлов промывают 50%-ным раствором уксусной кислоты до обесцвечивания элюата и высушивают. Ацетилирующую смесь готовят нз уксусного ангидрида и петролейного эфира (9 1 по объему), добавляя 1—2 канли концентрированного раствора Н2804 на каждые 100 мл, и тщательно перемешивают. Полоску бумаги опускают в ацетилирующую смесь на 40 мин, после чего бумагу промывают 3—4 раза, выдерживая ее по 15 мин в дистиллированной воде, затем высушивают при комнатной температуре. При дальнейшем использовании смеси время каждого последующего ацетилирования следует увеличивать на 5—10 мин. [c.305]
Ход определения. В коническую колбу отвесить с точностью до 0,01 г около 4 г жирных кислот. Навеску кипятить 1 ч в колбе с обратным холодильником с 25 мл 2 н. спиртового раствора едкого кали. Затем прибавить 25 мл воды и довести еще раз жидкость до кипения. Полученный мыльный раствор перенести из колбы в делительную воронку. Колбу обмыть два раза петролейный эфиром (каждый раз по 25 мл), сливая его в ту же делительную воронку. Взболтать. Выпустить нижний слой в другую делительную воронку и там обработать его опять 25 мл петролейного эфира. Обработку петролей-ным эфиром повторять до тех пор, пока нанесенная на фильтровальную бумагу капля эфира не перестанет оставлять следов жира. Соединенные эфирные вытяжки промыть три раза, добавляя каждый раз 15 мл 60-%-ного спирта, легко встряхивая до полного удаления остатков мыла промывная жидкость, разбавленная водой, не должна окрашиваться в присутствии фенолфталеина в розовый цвет. Слить эфирные вытяжки во взвешенную колбу для отгонки петролейного эфира. [c.179]
Прибор (рис. 40) состоит из фитильной лампочки 4, лампового стекла 3, абсорбера 1 и брызгоулови-теля 2. Перед определением лампочку 4 и фитиль 5 промывают петролейным эфиром и сушат. Все остальные детали тщательно моют и ополаскивают дистиллированной водой. В большой резервуар абсорбера насыпают до /3 его высоты чисто вымытые стеклянные бусы или отрезки стеклянных палочек в качестве насадки, улучшающей абсорбцию сернистого газа в поглотительном растворе. В абсорбер заливают из бюретки точно 10 мл раствора соды и 10 Л1Л дистиллированной воды. При анализе масел заливают 25 мл раствора- соды. Смазывают шлифы 6 и собирают прибор. К брызгоуловителю подключают систему вакуума, снабженную промежуточной буферной емкостью. На резиновые трубки между насосом и буферной емкостью и между этой емкостью и брызгоулови-телем надевают винтовые зажимы для регулирования просасывания [c.123]
Для определения мочевины отвешивают 0,5 г пробы и растворяют в 250 сма воды. Отбирают пипеткой 25 см3 в высокую широкогорлую склянку, емкостью около 100 см9, добавляют несколько капель индикатора метилрот и точно нейтрализуют, применяя 1/10 н. NaOH. Прибавляют 10 сма нейтральной уреазы и оставляют на один час, держа горло склянки закрытым. Вливают из бюретки отмеренный избыток 1/10 н. НС1 вставляют в склянку стеклянную трубку с шариком и маленькими дырочками на нижнем конце, и быстро продувают очищенный воздух в течение от 5 до 10 минут для удаления углекислоты. Две или три капли каприлового спирта или петролейного эфира прибавляют к раствору для предотвращения вспенивания. После продувания титруют до точной нейтрализации 1/10 н. NaOH. [c.113]
Определение постоянной зодного числа вискозиметра производят не реже 1 раза в 3 мес. Бели результаты проверки выходят за пределы 51 1 сек, то вискозиметр бракуется. Перед определением внутренний сосуд вискозиметра промывают последовательно петролейным эфиром, этиловым спиртом и дистиллированной водой, после чего высушивают его воздухом. Стержень при этом требуется новый, сухой, не имеющий на конце зазубрин и повреждений. Определение водного числа производят следующим образом. [c.196]
Растирают в ступке 50 г (0,21 М) хлоргидрата 5-хлорме-тил-8-хинолинола с 18,26 г (0,21 М) бикарбоната натрия и все содержимое переносят в круглодонпую колбу па 250 мл, в которую добавляют 125 мл (2,1 М) аллилового спирта (см. примечание I), Колбу соединяют с обратным холодильником и реакционную массу нагревают на масляной бане при кипении аллилового спирта до момента прекращения выделения углекислого газа, на что затрачивается 2,5—3 часа. После охлаждения реакционной массы образовавшийся желтый продукт отфильтровывают (см. примечание 2) и растворяют в 500 мл дистиллированной воды. Содержимое раствора нейтрализуют 5%-ным раствором аммиака до нейтральной реакции по универсальному индикатору. Выпавший белый осадок отфильтровывают и высушивают т. пл. 75°, выход равен 75%. После перекристаллизации из петролейного эфира (или октана, гептана) получают в виде белых игл 5-аллилоксиметил-8-хино-линол с температурой плавления 80—8Г и выходом 65% от теоретического (см. примечание 3). Элементарный анализ и определение непредельности подтверждают строение продукта. Вещество хорошо растворимо в бензоле, хлороформе, диоксане. [c.17]
Перимицин представляет собой золотисто-желтый аморфный порошок, не имеющий определенной температуры плавления, при нагревании темнеет с разложением. Метанольные растворы антибиотика имеют в ультрафиолете максимумы поглощения при 361, 383 и 406 нм. Перимицин растворяется в диметилформамиде, низших жирных кислотах, диметилсульфоксиде и метаноле при нагревании растворяется в низших -спиртах, пиридине, тетрагидрофуране, ацетоне и диоксане, но лишь при наличии воды не растворяется в воде, петролейном эфире, этилацетате и бензоле. [c.87]
Точную навеску около 0,2 г технической диацетонсорбозы помещают во взвешенный вместе с палочкой стакан емкостью около 50 мл и экстрагируют диацетоп-сорбозу при нагревании на водяной бане (при 40—50°) отдельными порциями (по 5 мл) петролейного эфира при тщательном растира.ции стеклянной палочкой содержимого стакана. Эфирные вытяжки сливают. Извлечение петролейным эфиром производят до тех пор, пока капля эфирного экстракта перестанет оставлять на часовом стекле пятно по испарении эфира. Остаток в стакане сушат в вакууме и взвешивают. Затем оста-Т01К растворяют в мерной колбе емкостью 100 мл о 20 мл воды, прибавляют 25 мл 60% раствора Н2504, колбу закрывают пробкой и оставляют на ночь. В дальнейшем поступают, как описано в методике определения диацетонсорбозы. [c.222]
Для определения кобальта экстрагируют кобальт из водного раствора раствором реагента в петролейно.М эфире [592]. Реагент позволяет обнаружить кобальт при разбавлении 1 10 000 000. Окраска устойчива и не изменяется несколько часов. При определении необходимо контролировать кислотность водного раствора, так как оптическая плотность зависит от pH. Наибольшая интенсивность окраски наблюдается при pH 3,8—4,4. Реагент взаимодействует также с солями паллг дия и железа (HI), образуя с ними соединения соответственн зеленого и коричневого цвета, которые также экстрагируются петролейным эфиром. Медь, ртуть, никель, цинк, железо (II) образуют с о-нитрозофенолом растворимые в воде окрашенные соединения, однако они, в отличие от комплексов кобальта, палладия и железа, не растворимы в петролейном эфире и поэтому не мешают. Влияние трехвалентного железа можно устранить применением цигратного буферного раствора, из раствора которого железо не экстрагируется раствором реагента в петролейном эфире. [c.142]
Свойства коры, важные для ее практического использования, определяются, кроме анатомического строения, химическим составом. Кора отличается от древесины поведением при набухании, меньшей анизотропностью, более низкими коэффициентами теплопередачи и механическими показателями [5, 57]. В коре в отличие от древесины присутствуют полифенолы и суберин, меньше массовая доля полисахаридов и больше доля экстрактивных веществ. Анализу подвергали кору различных видов, но из-за разных методик экстракции сравнение данных ограниченно. Массовая доля всех экстрактивных веществ в коре сосны ладанной (Pinus taeda), определенная последовательным экстрагированием петролейным эфиром, бензолом, этанолом, холодной и горячей водой, составляет 19,9 % [59], а при последовательном экстрагировании гексаном, бензолом, этиловым эфиром, этанолом, водой и 1 %-ным NaOH — 27,5 % [50]. При экстрагировании спиртобензольной смесью из коры сосны ладанной удаляется 18,3 % экстрактивных веществ [c.194]
Распределительную хроматографию фенолов, которая является по существу экстракционным процессом, чаще ведут на силикагеле. Стационарной фазой для разделения фенолов обычно служат метанол, вода элюэнтами — петролейный эфир, бензол, циклогексан, диэтиловый эфир, этилизоцианат, метанол или их смеси. Анализу подвергают узкие фракции (фенольную, крезольную, ксиленольную) [62, 63], а также более широкие, влючающие алкилфенолы, нафтолы, двухатомные фенолы [64, 65] и фракции смол коксования и полукоксования [64, 66]. Идентификацию и определение содержания фенолов в элюатах проводят колориметрическими и спектральными методами. Как показывают результаты анализов, в данном методе достигается довольно хорошее разделение фенолов по молекулярному весу и количеству гидроксильных групп в молекуле. Разделение изомеров обычно проходит не полностью. Замена силикагеля цеолитом [67] сокращает время анализа. [c.50]
Применение Н-соли в водных системах обусловлено ее растворимостью. Однако относительно чувствительности анализа К-соль не является оптимальной. Флороглюцин и нафтол обеспечивают более глубокую окраску, однако продукты реакции с ними нерастворимы в воде. Тем не менее реакцию азосочетания с этими реагентами можно провести, как описано для Н-соли, образуюш,ееся красяп ее веш ество извлечь бензолом, петролейным эфиром или четыреххлористым углеродом и провести колориметрический анализ органического слоя. Некоторые из красящих веществ— производные флороглюцина или нафтола — можно растворить, прибавляя щелочь (по окончании реакции сочетания, чтобы не вызвать разложения соли диазония). Щелочь образует соли с фенольными гидроксигруппами, придавая таким образом растворимость продуктам реакции. В этом случае возможно колориметрическое определение непосредственно водного раствора. [c.520]
Ввиду быстрого окисления метилового спирта в организме, количественное определение метилового спирта в перегоне внутренностей обыкновенно не может иметь места (см. стр. 80). Количественное определение в различных жидкостях (напитках, одеколонах и пр.) при достаточных количествах его, в отсутствии винного спирта, может быть, произведено по удельному весу дестиллата. Для этого дестиллат предварительно очищают от летучих кислот взбалтыванием со свежеосажденным углекислым кальцием от Э5би/ ных масел Спри исследовании одеколонов, настоек и прочих ароматны.х жидкостей) —извлечением последних при взбалтывании дестиллата (разбавленного равным объемом воды) с петролейным эфиром . [c.81]
Джонс и Маклахлан [160] при анализе различных материалов путем дистилляции в приборе Дина—Старка сравнивали результаты, получаемые при использовании бензола, толуола и петролейного эфира (т. кип. 90—100 °С). Результаты определения влаги в сливочном масле и маргарине оказались одинаковыми для этих растворителей и с правильностью 0,2—0,3% совпадали с данными, полученными при высушивании анализируемых материалов при 100 °С в воздушном сушильном шкафу с водяным обогревом и при 108 °С в воздушном сушильном шкафу. При анализе джемов, меда, пшеничных и солодовых экстрактов, бобов какао, подвергнутых ферментации, крахмала и мыла определяемое содержание воды было тем выше, чем выше температура кипения применяемого растворителя. Тем не менее все результаты укладываются в пределы широкого интервала значений, получаемых при сушке в воздушном сушильном шкафу при 100, 108 и 135 °С. Было показано, что результаты, получаемые методом дистилляции при анализе джема, меда и солодового экстракта, более надежны, чем данные, найденные при высушивании [160]. При использовании перечисленных выше переносящих агентов для моногидрата лактозы были найдены значения содержания воды соответственно 5,0 5,0 и 5,3%. Высушивание при 100 и 108 °С показало присутствие лишь 0,1 % влаги. [c.270]
Нефтепродукть . Метод азеотропной отгонки, по-вндимому, разработан именно на основе метода дистилляции нефтяных фракций вода при этом собирается в нижнем слое дистиллята. Одним из первых было сообщение Маркуссона [191 ] о применении толуола для анализа консистентных смазок. Дин и Старк [94] для определения влаги в нефтепродуктах использовали смесь 20% бензола и 80% ксилола или петролейный эфир (т. кип. 90—150 °С). Обычно для анализа нефтепродуктов применяют ксилол [4—6, 14, 300], толуол [4,5] или бензол [90]. Для определения влаги в пеках и ас-фальтах рекомендуется отгонка со смесью 20% бензола и 80% ксилола в аппарате Дина—Старка [14]. Воспроизводимость результатов при анализе асфальтовых эмульсий, содержащих 1— 50% воды, составляла 0,2—0,4%. При определении воды в минеральных маслах Фукс [117] использовал метод отгонки с бензолом. Для более четкого выявления капель воды в органическом слое он добавлял в ловушку 1—2 мл концентрированного раствора асфальта в бензоле. При этом на фоне окрашенного бензола были хорошо видны бесцветные капли воды. Их удаляли легким постукиванием или осторожным нагреванием приемника. В официальном методе ASTM для определения воды в нефтепродуктах и других битумных материалах [4—6] применяют приборы Дина—Старка (см. рис. 5-1 и 5-2). [c.275]
Широкое применение при анализе мыл находит отгонка воды с ксилолом. Однако Траслер [290] установил, что использование ксилола приводит к существенным ошибкам при анализе мыл, в состав которых входит глицерин. При использовании бензола и толуола, кипящих при более низких температурах, ошибки весьма незначительны. При сравнении методов анализа мыл, содержащих глицерин и 25—60% воды, показано, что результаты, получаемые при использовании ксилола, на 0,5—1% выше, чем при отгонке воды с толуолом. В тех случаях, когда анализируемые мыла не содержат глицерин, расхождение результатов составляет до 0,1%. При проведении анализа в перегонный сосуд добавляют безводный хлорид бария в качестве агента, препятствующего пенообразованию [289]. Принятый АОАС метод определения влаги в мылах и в стабилизованных мылами эмульсиях минеральных масел [18] предусматривает отгонку воды с толуолом. Для предотвращения пенообразования рекомендуется добавлять небольшое количество канифоли. Для анализа мыл, содержащих более 1 % силиката натрия, глицерина, ароматических веществ или солей аммония применяют петролейный эфир (т. кип. 100—120 °С) [59]. Для анализа моющих средств, содержащих фосфатные структуро-образователи, рекомендуется петролейный эфир (т. кип. 140— 160 °С) [59]. [c.282]
Метод определения воды по критической температуре растворения (метод КТР) был применен для анализа смесей спиртов с углеводородами [22, 41, 144, 145, 151, 192]. Плит [ 145], рассматривая ряд методов определения воды в моторных топливах, отмечал, что примесь 0,2 % воды в системе метанол — циклогексан вызывает повышение КТР на 1,35 °С. Составлены таблицы критических температур растворения воды в смесях метанол—циклогексан и этанол—бициклогексил [192]. При определении воды в смесях этанол—углеводород Крисмер [41 ] использует в качестве водонерастворимой фазы смесь керосина и петролейного эфира, Ботсет [c.541]
chem21.info