Реферат: Синтез диэтилового эфира малоновой кислоты. Свойства и основные методы получения сложных эфиров. Диэтиловый эфир малеиновой кислоты
Диэтиловый эфир - малеиновая кислота
Диэтиловый эфир - малеиновая кислота
Cтраница 1
Диэтиловый эфир малеиновой кислоты был получен нагреванием кислоты с-абсолютным спиртом в присутствии серной кислоты. [1]
Двойная связь диэтилового эфира малеиновой кислоты полностью насыщается как газообразной, так и жидкой азотноватой окисью. [2]
Показано, что диэтиловый эфир малеиновой кислоты изомери-зуется в диэтиловый эфир фумаровой кислоты как под влиянием газообразной, так и жидкой азотноватой окиси. [3]
Действием жидкой азотноватой окиси на диэтиловый эфир малеиновой кислоты получен псевдонитрозит и, весьма вероятно, диазо-тистый эфир. [4]
Оксазол смешивают с двумя молями диэтилового эфира малеиновой кислоты, нагревают при 110 С в течение 2 ч и после обработки аддукта соляной кислотой получают диэтиловый эфир 2-метил - 3-окси - 4 5-дикарбоксипиридина с выходом 85 %, температура плавления 140 - 144 С ( лит. Этот диэфир восстанавливают литийалюмогидридом ( LiAlh5) в пиридоксин. Дицианпиридин превращают в пиридоксин гидрогенизацией на палладиевом катализаторе с получением 2-метил - 3-окси - 4 5-диаминометилпиридина ( выход 70 %, температура плавления 290 С, лит. Кондратьевой; однако отсутствие публикаций по технологии метода не позволяет отдать ему предпочтение перед другими методами. [5]
Присоединение электрохимически генерируемых этильных радикалов к диэтиловому эфиру малеиновой кислоты. [6]
СНз) с кротоновой кислотой, диэтиловым эфиром малеиновой кислоты, олеиновой и абиетиновой кислотами; в двух последних случаях строение аддуктов не установлено. [7]
ТУПС [1878] подвергал фракционированной перегонке 3 л диэтилового эфира малеиновой кислоты ( СУДЯ по результатам омыления 99 25 % - ного) на колонке с 20 тарелками ( типа Perm State), заполненной одновитковыми стеклянными спиралями диаметром 3 мм. [8]
Нагревают три моля альдегида и 1 моль диэтилового эфира малеиновой кислоты 24 час с обратным холодильником при температуре 82 - 85, причем вначале и спустя 8 час прибавляют по 1 % перекиси дибензоила. Чтобы получить хороший выход, необходимо строго выдерживать заданную температуру. [10]
Нагревают три моля альдегида и 1 моль диэтилового эфира малеиновой кислоты 24 час с обратным холодильником при температуре 82 - 85, причем вначале и спустя 8 час прибавляют по 1 % перекиси дибензоила. Чтобы получить хороший выход, необходимо строго выдерживать заданную температуру. [12]
Представлял интерес вопрос о том, может ли изомеризоваться диэтиловый эфир малеиновой кислоты в эфир фумаровой кислоты и обратно под влиянием азотноватой окиси. [13]
Очистку сырой малеиновой кислоты, полученной окислением бензола или нафталина, перекристаллизацией из растворителей ( например из диэтилового эфира малеиновой кислоты или из смесей сложных эфиров малеиновой кислоты с гидрированными многоядерными: ароматическими углеводородами) описал. [14]
Страницы: 1 2
www.ngpedia.ru
Реферат - Синтез диэтилового эфира малоновой кислоты Свойства и основные методы получения сложных эфиров
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования
Самарский государственный технический университет
Кафедра: «Органическая химия»
“СИНТЕЗ ДИЭТИЛОВОГО ЭФИРА МАЛЕИНОВОЙ КИСЛОТЫ”
Курсовая работа
Выполнил:
Руководитель:
Самара, 2007 г.
Содержание
1. Введение
1.1. Свойства диэтилового эфира малеиновой кислоты
1.2. Практическое применение
1.3. Методика синтеза
2. Литературный обзор
2.1. Двухосновные (дикарбоновые) кислоты
2.2. Реакция этерификации
2.3. Механизм этерификации
3. Выводы
Список литературы
1. Введение
Свойства диэтилового эфира малеиновой кислоты
Диэтиловый эфир малеиновой кислоты, диэтилмалеат, этилмалеат C2H5OOCCH=CHCOOC2H5. Температура замерзания: ~-11.5°C, температура кипения: 225.3°C, дипольный момент: 2.54 Дебай, диэлектрическая проницаемость: 8.58 при 230С, плотность: 1.0687 при 20°С, г/мл, показатель преломления: 1.4400 при 20°С. Легковоспламеняющаяся жидкость
Практическое применение
Основная область применения диэтилмалеата – использование в качестве органического растворителя.
Органические растворители используются как в аналитической химии, так и в производстве. Среди потребителей органических растворителей – лаборатории и научно-исследовательские организации, предприятия нефтехимической, фармацевтической, парфюмерной, пищевой, электронной и оборонной промышленности.
Кроме того, широкое применение органические растворители, в частности диэтилмалеат, находят в лакокрасочной промышленности.
Постоянное ужесточение законодательства по охране окружающей среды привело к значительному вытеснению в последние годы традиционных красок на органических растворителях более экологически чистыми — водорастворимыми красками. Однако органоразбавляемые краски довольно часто используются в строительстве благодаря высокому качеству покрытий и относительному удобству применения. По разным оценкам их доля в общем объеме потребления строительных красок стабилизировалась на уровне 20-30%.
В настоящее время органоразбавляемые краски включены в программы большинства ведущих производителей лакокрасочных материалов. Чаще всего в качестве растворителя в современных органоразбавляемых красках применяют относительно низкотоксичный уайт-спирит, хотя иногда применяют и токсичные растворители (например, сольвент и ксилол). Кроме токсичности следствием применения в составе красок органических растворителей является их горючесть, а также характерный, часто достаточно сильный запах.
С появлением водоразбавляемых красок принято считать, что краски на органических растворителях имеют по сравнению ними всего два неоспоримых преимущества. Первое преимущество — возможность применения при отрицательных температурах (по материалам некоторых производителей до -20 0С). Второе преимущество — состоит в том, что свеженанесенное, еще не стабилизированное покрытие не может быть повреждено дождем.
Оба эти преимущества позволяют существенно расширить сезонность поведения работ, продлив ее на весну и осень. Теоретически возможно применение таких красок и в зимний период, однако это связано с рядом технологических сложностей, связанных с необходимостью предварительного оттаивания и осушения подложки.
Достаточно мощный импульс к использованию органоразбавляемых красок дало применения в качестве пленкообразователя специальный термопластиковой акриловой смолы Плиолит (PLIOLIT — торговая марка The Goodyear Tire & Rubber Co, USA).
Естественно краски на плиолитовых смолах обладают всеми перечисленными выше преимуществами органоразбавляемых красок, что однако не является главным. Главное же состоит в том, что они образуют достаточно хорошее покрытие, сравнимое с теми, которые можно получить с применением водоразбавляемых красок самых последних поколений.
На Российском рынке краски на основе плиолитовых смол представлены следующими фирмами: Alpa (Франция), Marshall (группа Akzo Nobel, Турция), Murolite (Швеция), Soframap (Франция)
Методика синтеза
/>
Малеиновая кислота 29 г (0,25 г-моль)
Этиловый спирт 96%-ный 32 г
Бензол 20 мл
Серная кислота (d=1,84). Бикарбонат натрия
/>
Приборы для проведения синтезов с азеотропной отгонкой воды: а– с холодильником Либиха, б– с шариковым холодильником, 1– реакционная колба, 2– двурогий форштосс, 3– капельная воронка, 4– «ловушка» для воды, 5– обратный холодильник.
В круглодонной колбе, снабженной обратным холодильником и ловушкой для воды, смешивают 29 г малеиновой кислоты, 32 г этилового спирта, 1,5 мл концентрированной серной кислоты и 20 мл бензола. Смесь кипятят на водяной бане или колбонагревателе до прекращения выделения воды, охлаждают, переносят в делительную воронку и промывают водой, последовательно водным раствором бикарбоната натрия и еще раз водой. После этого отгоняют растворитель, который захватывает с собой и следы воды. Остаток перегоняют из колбы с дефлегматором.
Выход диэтилмалеата 34 г (79% теоретического), температура кипения 123°Спри 12 мм рт. ст., />.
Литературный обзор.
Двухосновные (дикарбоновые) кислоты
Диэтилмалеат является сложным эфиром двухосновной малеиновой кислоты. Чтобы иметь представление о свойствах и структуре данного эфира, рассмотрим кратко этот класс органических соединений.
Общая формула этих кислот НООС–(СН2)n–СООН. Тривиальные названия имеют только первые члены ряда:
№
Название кислоты
Температура плавления
Растворимость, г/100 г Н2О при 20° С
Щавелевая
179,5
8,0
1
Малоновая
135,6
73,5
2
Янтарная
188 ,0
5,8
3
Глутаровая
97,5
63,9
4
Адипиновая
153,0
1,6
5
Пимелиновая
105,
5,0
6
Пробковая
144,0
0,16
7
Азелаиновая
106,5
0,24
8
Себациновая
134,5
0,1
9
Нонандикарбоновая
111
10
Декандикарбоновая
128
11
Брассиловая
113
12
Додекандикарбоновая
126
13
Тридекандикарбоновая
113,5
14
Тапсиевая
125
--PAGE_BREAK--
Зависимость температуры плавления от числа атомов углерода в молекулах представляет собой «пилу» с еще более острыми зубцами. Объяснение такое же, как и для монокарбоновых кислот: разное строение кристаллической решетки для четных и нечетных членов ряда. Для первых семи членов ряда наблюдается также сильное альтернирование величин растворимости кислот в воде. Понятно, что это свойство также связано с кристаллической решеткой: чем она прочнее, тем меньше растворимость.
Простейшая двухосновная щавелевая кислота содержит две соединенные карбоксильные группы НООС–СООН. Ее соли и эфиры называются оксалатами (от греч. oxys – кислый). Эта кислота известна с 17 в., она содержится (в виде калиевой соли) в щавеле (ее там 0,36%), откуда и получила свое название. Есть она и в других овощах и плодах: в шпинате ее 0,32%, в томатах – 0,06%. Избыток щавелевой кислоты может нарушать обмен веществ в организме, способствуя отложению нерастворимого оксалата кальция. Поэтому врачи рекомендуют ограничить потребление продуктов с повышенным содержанием этой кислоты.
Щавелевая кислота – одна из самых сильных органических кислот: при диссоциации по первой ступени она значительно сильнее уксусной. Она образует хорошо растворимые комплексные соединения со многими металлами, что используют для очистки металлов от ржавчины, для выведения ржавых пятен с тканей, сантехнических изделий и т.д. Например, ржавое пятно на белой ткани, смоченное раствором щавелевой кислоты, исчезает прямо на глазах.
Диэтиловый эфир малоновой кислоты (от лат. malum – яблоко) широко применяется в органических ситезах; химики называют его просто «малоновым эфиром». От этого же корня происходят названия непредельной малеиновой кислоты (цис-НООС–СН=СН–СООН) и производных яблочной кислоты – малатов. Интересно название транс-изомера малеиновой кислоты – фумаровой (от лат. fumus – дым). Эта кислота была обнаружена в растении Fumaria officinalis (дымянка), которое в античные времена сжигали, чтобы дымом отогнать злых духов.
Янтарная кислота была получена еще в 17 в. перегонкой янтаря, ее соли и эфиры назваются сукцинатами (лат. succinum – янтарь). Глутаровая кислота впервые была получена из глутаминовой аминокислоты, а та получила свое название от лат. gluten – клей, поскольку была найдена в клейковине пшеницы. Адипиновая кислота образуется при окислении жиров и получила название от лат. adeps – жир, сало. Эту кислоту синтезируют в промышленных масштабах, так как она является исходным веществом для производства полиамидных волокон (найлон-6,6) и смол. Кстати, название этого полимера происходит от первых букв двух городов – New York, London и числа атомов углерода в остатках адипиновой кислоты и гексаметилендиамина h3N –(Ch3)6 –Nh3, которые соединены поочередно в полимерную цепь. Название пимелиновой кислоты происходит от греч. pimelos – жир, субериновой (пробковой) кислоты – от лат. suber – пробка, себациновой кислоты – от лат. sebum – сало. Азелаиновая кислота была получена действием азотной кислоты на касторовое масло. Соответственно в ее названии можно найти «азо» и греч. elaion – масло.
Двухосновные кислоты с числом атомов углерода более 10 имеют обычно систематические названия. Но есть и исключения: брассиловая кислота была найдена в масле растений семейства Brassica; тапсиевая – в растении тапсия с греческого острова Тапсос, которое употреблялось в древности как лекарственное; японовая НООС–(СН2)19–СООН – выделена из высушенного сока некоторых акаций и пальм, растущих в Юго-Восточной Азии (раньше это вещество называли «японской землей»)
Реакция этерификации
Основным способом получения сложных эфиров карбоновых кислот является реакция этерификации. Диэтилмалеат не является исключением. Этерификация – практически главнейший способ получения данного эфира двухосновной кислоты. Рассмотрим основные свойства реакции этерификации.
Итак, реакцией этерификации называется взаимодействие спиртов с карбоновыми кислотами, приводящее к образованию сложных эфиров:
/>
В этой реакции молекула спирта выступает в роли нуклеофильного агента, атакующего бедный электронами углеродный атом карбонильной группы.
Реакции этерификации обратимы и, следовательно, ограничены состоянием равновесия. Превращение эквимолекулярных количеств кислоты и спирта в теоретически вычисленное количество сложного эфира по причине обратимости реакции невозможно. В результате реакции образуется некоторое максимальное количество эфира (которое всегда ниже теоретического) и остаются непрореагировавшие спирт и кислота. Например, при нагревании с обратным холодильником эквимолекулярных количеств уксусной кислоты и этилового спирта в реакцию вступает лишь 2/3 г-мол каждого компонента, поэтому максимальный выход эфира в этих условиях составляет лишь 2/3 теоретического, т. е. 66,7%.
/>
По мере того как кислота и спирт реагируют друг с другом и происходит накопление продуктов их взаимодействия (эфира и воды), скорость обратной реакции, вначале незначительная, возрастает. При этом скорость прямой реакции постепенно уменьшается. Наконец, наступает динамическое равновесие, когда в единицу времени в сложный эфир превращается столько же молекул кислоты и спирта, сколько молекул сложного эфира распадается на кислоту и спирт. Одинаковой скоростью этих противоположно протекающих процессов обусловлен постоянный состав системы. Поскольку скорость бимолекулярной реакции пропорциональна произведению концентраций реагирующих веществ, мы можем для скоростей прямой и обратной реакций написать уравнения:
/>
где v1— скорость реакции этерификации; v2— скорость реакции гидролиза; К1и К2— константы скорости обеих реакций; Ск, Сс, Сэи Св— концентрации реагирующих и получающихся веществ (кислоты, спирта, эфира, воды).
В состоянии равновесия скорости реакций, протекающих в противоположных направлениях, равны, т. е. V1= V2. Тогда К1СкСс= КгСэСвили:
/>
Частное К2/К1является константой равновесия и обозначается буквой К.
Из полученного уравнения следует, что в состоянии равновесия отношение произведений концентраций реагирующих веществ обратно отношению констант скоростей реакций. В случае реакции образования уксусноэтилового эфира в состоянии равновесия, как упомянуто выше, в реакционной смеси содержится по 1/3 моля кислоты и спирта и по 2/3 моля эфира и воды. Поэтому
/>
Однако можно изменить состояние равновесия и повысить выход сложного эфира, увеличивая концентрацию спирта (или кислоты). Например, если взять уксусную кислоту и спирт в молярном отношении, равном 1:2, выход эфира (из расчета на кислоту) повышается до 85%. Действительно, пусть концентрация эфира в состоянии равновесия (в молях) будет равна х, т. е. Сэ= х. Тогда и Св= х. Концентрация кислоты Ск= 1—х, концентрация спирта Сс= 2 — х. Следовательно,
/>
После решения этого уравнения находим, что х = 0,85 моля, то есть выход эфира равен 85% теоретического.
Часто применяется и другой способ смещения равновесия в сторону большего выхода сложного эфира — удаление сложного эфира или воды из сферы реакции. Легко можно видеть, что уменьшение концентраций эфира или воды влечет уменьшение концентраций спирта и кислоты, поскольку величина константы равновесия К при данной температуре неизменна. Так, в случае получения низкокипящих сложных эфиров (например, уксусно-этилового с температурой кипения 77°С) в ходе реакции отгоняют эфир из реакционной колбы. При получении высококипящих сложных эфиров (например, уксуснобутилового с температурой кипения 125°С или уксусноизоамилового с температурой кипения 142°С) удобнее отгонять воду в процессе реакции. Вода в этом случае отгоняется в виде азеотропа с парами соответствующего спирта. При конденсации паров в холодильнике происходит расслоение этих ограниченно смешивающихся жидкостей и вода, как более тяжелая, собирается на дне поставленной на пути конденсата «ловушки» (см. рис. 27). Азеотропную отгонку воды можно использовать и в случае этерификации кислот этиловым или пропиловым спиртом, которые в жидкой фазе смешиваются с водой во всех отношениях. В этом случае для отделения воды от сконденсировавшегося в холодильнике спирта в реакционную смесь приходится добавлять третий компонент, образующий с водой и спиртом нераздельно кипящую смесь, но в жидкой фазе с водой не смешивающийся. Его роль состоит в том, что он экстрагирует из конденсата спирт и возвращает его в реакционный сосуд. В качестве такого компонента могут использоваться бензол, хлороформ, четыреххлористый углерод и некоторые другие жидкости, но из перечисленных только бензол можно использовать в «ловушках». Хлороформ и четыреххлористый углерод обладают большей плотностью, чем вода, и для отделения воды от реакционной смеси в случае использования этих жидкостей требуется «ловушка» другой конструкции.
продолжение --PAGE_BREAK--При комнатной температуре реакция протекает очень медленно. При смешении эквимолярных количеств спирта и кислоты для достижения равновесных концентраций требуется до 16 лет. Повышение температуры ускоряет реакцию (так, в случае взаимодействия этилового спирта с уксусной кислотой при 110°Сравновесие достигается через 10 дней, а при (155°С— через несколько часов).
Особенно сильное ускорение реакции этерификации достигается применением катализаторов — водородных ионов, получающихся при диссоциации сильных минеральных кислот. В качестве катализаторов чаще всего используются концентрированная серная кислота или сухой хлористый водород, ток которого пропускается через реакционную смесь. Найдено, что скорость реакции возрастает с увеличением количества катализатора; однако известно также, что добавка 0,01% серной кислоты достаточна для образования этилацетата из спирта и уксусной кислоты. Следует иметь в виду, что катализаторы повышают скорость реакции этерификации, но не могут вызывать сдвига равновесия.
Карбоновые кислоты, как видно из вышесказанного, реагируют со спиртами относительно медленно. Это объясняется слабой активностью карбонильной группы в кислотах по отношению к нуклеофильным агентам по сравнению с активностью той же группы в ангидридах и хлорангидридах кислот, поскольку +М-эффект гидроксильной группы приводит к уменьшению положительного заряда карбонильного углерода
/>
Скорость этерификации карбоновой кислоты тем выше, чем больше положительный заряд карбонильного углерода. Величина δ+ на углероде карбоксильной группы зависит от характера радикала кислоты. Электронодонорные группы, связанные с карбоксилом, понижают дробный положительный заряд (по сравнению с зарядом в муравьиной кислоте) и тем препятствуют взаимодействию кислоты с нуклеофилом; электроноакцепторные заместители, напротив, делают кислоту более реакционноспособной. Поэтому кислоты типа трихлоруксусной, щавелевой, муравьиной быстро реагируют со спиртами даже без добавок минеральной кислоты-катализатора, а ароматические кислоты, особенно те, которые в ароматическом ядре содержат электронодонорные заместители, взаимодействуют со спиртом значительно труднее и требуют больших количеств катализатора.
Сильное влияние на скорость реакции этерификации оказывают также пространственные факторы. С увеличением объема связанных с карбоксилом углеводородных радикалов и с повышением объема этерифицируемых спиртов скорость этерификации уменьшается. Среди спиртов одного молекулярного веса быстрее всего взаимодействуют с кислотами первичные, медленнее — третичные спирты.
Реакцию этерификации можно проводить и в паровой фазе над твердыми катализаторами. Пары спирта и кислоты при 280—300° С пропускают через трубку с катализатором (ThO2 или TiO2). Выходы сложных эфиров в этом случае такие же, как и при реакциях в гомогенной фазе.
Аминокислоты образуют сложные эфиры при взаимодействии со спиртами в присутствии сухого хлористого водорода. Роль хлористого водорода здесь не ограничивается катализом реакции или сдвигом равновесия за счет связывания воды. В присутствии хлористого водорода аминокислота, находившаяся ранее в форме внутренней соли, превращается в хлористоводородную соль аминокислоты, причем карбоксильная группа из неактивной формы аниона переходит в реакционноспособную форму —СООН:
/>
В результате этерификации в этих условиях эфиры также получаются в виде солей. Например, из аминоуксусной кислоты (гликоколя) и абсолютного этилового спирта образуется хлористоводородная соль эфира гликоколя
/>
Свободный эфир из соли можно получить, удаляя хлористый водород окисью серебра:
/>
Механизм этерификации
Роль катализатора заключается в протонировании карбонильного кислорода: при этом карбонильный атом углерода становится более положительным и более «уязвимым» по отношению к атаке нуклеофильного агента, которым является молекула спирта. Образующийся вначале катион (VIII) присоединяет молекулу спирта за счет неподеленных электронов кислородного атома, давая катион (IX):
/>
Далее катион (IX) отщепляет молекулу воды, превращаясь в катион сложного эфира (X):
/>
Катион (X) в результате отщепления протона образует молекулу сложного эфира:
/>
Использование метода «меченых атомов» дало возможность решить вопрос о месте разрыва связей при реакции этерификации. Оказалось, что обычно молекула воды образуется из гидроксила кислоты и водорода спирта. Следовательно, в молекуле кислоты разрывается связь между ацилом и гидроксилом, а в молекуле спирта — связь водорода с кислородом. Такой именно вывод следует из результатов работы по этерификации бензойной кислоты метанолом, содержащим тяжелый изотоп кислорода О18. Полученный сложный эфир содержал в своем составе указанный изотоп кислорода:
Присутствие О18установлено сжиганием образца эфира и анализом образующихся продуктов сгорания (CO2и Н2О) на присутствие тяжелого изотопа кислорода.
Гидролиз сложных эфиров представляет собой реакцию, обратную реакции их образования. Гидролиз может быть осуществлен как в кислой, так и в щелочной среде. Для кислого гидролиза сложных эфиров справедливо все, что было сказано выше применительно к реакции этерификации, об обратимости и механизме процесса, о методах смещения равновесия. Щелочной гидролиз сложных эфиров проходит через следующие стадии:
/>
Он является процессом необратимым, поскольку богатый электронами анион кислоты не способен взаимодействовать с нуклеофильной молекулой спирта.
Практически щелочной гидролиз сложных эфиров проводят в присутствии едких щелочей КОН, NaOH, а также гидроокисей щелочноземельных металлов Ва(ОН)2, Са(ОН)2Образующиеся при гидролизе кислоты связываются в виде солей соответствующих металлов, поэтому гидроокиси приходится брать по крайней мере в эквивалентном отношении со сложным эфиром. Обычно используют избыток основания. Выделение кислот из их солей осуществляется с помощью сильных минеральных кислот.
В качестве растворителя основания для реакции гидролиза чаще всего применяют воду, которая, однако, не растворяет сложный эфир. Реакция идет на поверхности раздела двух фаз и требует поэтому хорошего перемешивания. Иногда реакцию бывает целесообразно проводить в гомогенной среде, используя в качестве растворителя водный спирт. При этом, однако, нужно иметь в виду, что для выделения кислоты перед подкислением раствора спирт необходимо удалить (отогнать).
Список литературы
Общая органическая химия. Карбоновые кислоты и их производные. Том 4. М., Химия, 1983, 729с.
Шабаров Ю.С. Органическая химия: В 2-х кн. — М.: Химия, 1994.- 848 с.
Петров А.А., Бальян Х.В., Трощенко А.Т. Органическая химия. – М.: Высш. шк., 1973. — 623 с.
Препаративная органическая химия. Изд. 2-е, М., Госхимиздат, 1964.
Богословский Б.Н., Казакова З.С. Скелетные катализаторы, их свойства и применение в органической химии. М., Госхимиздат, 1957.
Голодников Г.В. Практические работы по органическому синтезу. Л., Изд-во ЛГУ, 1966, 697с.
Дорофеенко Г.Н., Жданов Ю.А., Дуленко В.И. и др. Хлорная кислота и ее соединения ворганическом синтезе. Ростов, изд-во Ростовского ун-та, 1965.
Терней А. Современная органическая химия: В 2 т. — М.: Мир, 1981. — Т.1 — 670 с; Т.2 — 615 с.
Лабораторные работы по органической химии. Изд. 3-е. М., Высшая школа, 1974.
Храмкина М.Н. Практикум по органическому синтезу. Изд. 4-ое, Л., Химия. 1977.
В. Ф. Травень. Органическая химия. Том 1. – М.: Академкнига, 2004, — 708 с.
Голодников Г.В., Низовкина Т.В., Рыскальчук А.Т. Практикум по органическому синтезу. Л., Изд-во ЛГУ, 1967.
Крешков А.П., Курбатов И.Н. Лабораторные работы по синтезу и анализу органических соеднений. М., изд-во Артиллерийского ордена Ленина академии Красной армии им. Дзержинского, 1940.
www.ronl.ru
Реферат - Синтез диэтилового эфира малоновой кислоты. Свойства и основные методы получения сложных эфиров
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования
Самарский государственный технический университет
Кафедра: «Органическая химия»
“СИНТЕЗ ДИЭТИЛОВОГО ЭФИРА МАЛЕИНОВОЙ КИСЛОТЫ”
Курсовая работа
Выполнил:
Руководитель:
Самара, 2007 г.
Содержание
1. Введение
1.1. Свойства диэтилового эфира малеиновой кислоты
1.2. Практическое применение
1.3. Методика синтеза
2. Литературный обзор
2.1. Двухосновные (дикарбоновые) кислоты
2.2. Реакция этерификации
2.3. Механизм этерификации
3. Выводы
Список литературы
1. Введение
1.1. Свойства диэтилового эфира малеиновой кислоты
Диэтиловый эфир малеиновой кислоты, диэтилмалеат, этилмалеат C2 H5 OOCCH=CHCOOC2 H5. Температура замерзания: ~-11.5°C, температура кипения: 225.3°C, дипольный момент: 2.54 Дебай, диэлектрическая проницаемость: 8.58 при 230С, плотность: 1.0687 при 20°С, г/мл, показатель преломления: 1.4400 при 20°С. Легковоспламеняющаяся жидкость
Основная область применения диэтилмалеата – использование в качестве органического растворителя.
Органические растворители используются как в аналитической химии, так и в производстве. Среди потребителей органических растворителей – лаборатории и научно-исследовательские организации, предприятия нефтехимической, фармацевтической, парфюмерной, пищевой, электронной и оборонной промышленности.
Кроме того, широкое применение органические растворители, в частности диэтилмалеат, находят в лакокрасочной промышленности.
Постоянное ужесточение законодательства по охране окружающей среды привело к значительному вытеснению в последние годы традиционных красок на органических растворителях более экологически чистыми — водорастворимыми красками. Однако органоразбавляемые краски довольно часто используются в строительстве благодаря высокому качеству покрытий и относительному удобству применения. По разным оценкам их доля в общем объеме потребления строительных красок стабилизировалась на уровне 20-30%.
В настоящее время органоразбавляемые краски включены в программы большинства ведущих производителей лакокрасочных материалов. Чаще всего в качестве растворителя в современных органоразбавляемых красках применяют относительно низкотоксичный уайт-спирит, хотя иногда применяют и токсичные растворители (например, сольвент и ксилол). Кроме токсичности следствием применения в составе красок органических растворителей является их горючесть, а также характерный, часто достаточно сильный запах.
С появлением водоразбавляемых красок принято считать, что краски на органических растворителях имеют по сравнению ними всего два неоспоримых преимущества. Первое преимущество — возможность применения при отрицательных температурах (по материалам некоторых производителей до -20 0С). Второе преимущество — состоит в том, что свеженанесенное, еще не стабилизированное покрытие не может быть повреждено дождем.
Оба эти преимущества позволяют существенно расширить сезонность поведения работ, продлив ее на весну и осень. Теоретически возможно применение таких красок и в зимний период, однако это связано с рядом технологических сложностей, связанных с необходимостью предварительного оттаивания и осушения подложки.
Достаточно мощный импульс к использованию органоразбавляемых красок дало применения в качестве пленкообразователя специальный термопластиковой акриловой смолы Плиолит (PLIOLIT — торговая марка The Goodyear Tire & Rubber Co, USA).
Естественно краски на плиолитовых смолах обладают всеми перечисленными выше преимуществами органоразбавляемых красок, что однако не является главным. Главное же состоит в том, что они образуют достаточно хорошее покрытие, сравнимое с теми, которые можно получить с применением водоразбавляемых красок самых последних поколений.
На Российском рынке краски на основе плиолитовых смол представлены следующими фирмами: Alpa (Франция), Marshall (группа Akzo Nobel, Турция), Murolite (Швеция), Soframap (Франция)
Малеиновая кислота… 29 г (0,25 г-моль)
Этиловый спирт 96%-ный… 32 г
Бензол… 20 мл
Серная кислота ( d = 1,84). Бикарбонат натрия
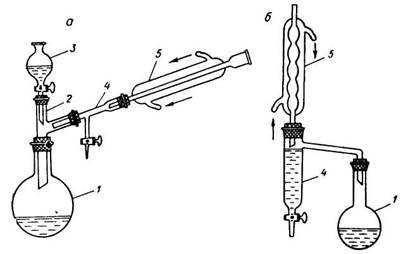
Приборы для проведения синтезов с азеотропной отгонкой воды: а – с холодильником Либиха, б – с шариковым холодильником, 1 – реакционная колба, 2 – двурогий форштосс, 3 – капельная воронка, 4 – «ловушка» для воды, 5 – обратный холодильник.
В круглодонной колбе, снабженной обратным холодильником и ловушкой для воды, смешивают 29 г малеиновой кислоты, 32 г этилового спирта, 1,5 мл концентрированной серной кислоты и 20 мл бензола. Смесь кипятят на водяной бане или колбонагревателе до прекращения выделения воды, охлаждают, переносят в делительную воронку и промывают водой, последовательно водным раствором бикарбоната натрия и еще раз водой. После этого отгоняют растворитель, который захватывает с собой и следы воды. Остаток перегоняют из колбы с дефлегматором.
Выход диэтилмалеата 34 г (79% теоретического), температура кипения 123°С при 12 мм рт. ст., .
Диэтилмалеат является сложным эфиром двухосновной малеиновой кислоты. Чтобы иметь представление о свойствах и структуре данного эфира, рассмотрим кратко этот класс органических соединений.
Общая формула этих кислот НООС–(СН2 )n –СООН. Тривиальные названия имеют только первые члены ряда:
№ | Название кислоты | Температура плавления | Растворимость, г/100 г Н2 О при 20° С |
Щавелевая | 179,5 | 8,0 | |
1 | Малоновая | 135,6 | 73,5 |
2 | Янтарная | 188 ,0 | 5,8 |
3 | Глутаровая | 97,5 | 63,9 |
4 | Адипиновая | 153,0 | 1,6 |
5 | Пимелиновая | 105, | 5,0 |
6 | Пробковая | 144,0 | 0,16 |
7 | Азелаиновая | 106,5 | 0,24 |
8 | Себациновая | 134,5 | 0,1 |
9 | Нонандикарбоновая | 111 | |
10 | Декандикарбоновая | 128 | |
11 | Брассиловая | 113 | |
12 | Додекандикарбоновая | 126 | |
13 | Тридекандикарбоновая | 113,5 | |
14 | Тапсиевая | 125 |
Зависимость температуры плавления от числа атомов углерода в молекулах представляет собой «пилу» с еще более острыми зубцами. Объяснение такое же, как и для монокарбоновых кислот: разное строение кристаллической решетки для четных и нечетных членов ряда. Для первых семи членов ряда наблюдается также сильное альтернирование величин растворимости кислот в воде. Понятно, что это свойство также связано с кристаллической решеткой: чем она прочнее, тем меньше растворимость.
Простейшая двухосновная щавелевая кислота содержит две соединенные карбоксильные группы НООС–СООН. Ее соли и эфиры называются оксалатами (от греч. oxys – кислый). Эта кислота известна с 17 в., она содержится (в виде калиевой соли) в щавеле (ее там 0,36%), откуда и получила свое название. Есть она и в других овощах и плодах: в шпинате ее 0,32%, в томатах – 0,06%. Избыток щавелевой кислоты может нарушать обмен веществ в организме, способствуя отложению нерастворимого оксалата кальция. Поэтому врачи рекомендуют ограничить потребление продуктов с повышенным содержанием этой кислоты.
Щавелевая кислота – одна из самых сильных органических кислот: при диссоциации по первой ступени она значительно сильнее уксусной. Она образует хорошо растворимые комплексные соединения со многими металлами, что используют для очистки металлов от ржавчины, для выведения ржавых пятен с тканей, сантехнических изделий и т.д. Например, ржавое пятно на белой ткани, смоченное раствором щавелевой кислоты, исчезает прямо на глазах.
Диэтиловый эфир малоновой кислоты (от лат. malum – яблоко) широко применяется в органических ситезах; химики называют его просто «малоновым эфиром». От этого же корня происходят названия непредельной малеиновой кислоты (цис -НООС–СН=СН–СООН) и производных яблочной кислоты – малатов. Интересно название транс -изомера малеиновой кислоты – фумаровой (от лат. fumus – дым). Эта кислота была обнаружена в растении Fumaria officinalis (дымянка), которое в античные времена сжигали, чтобы дымом отогнать злых духов.
Янтарная кислота была получена еще в 17 в. перегонкой янтаря, ее соли и эфиры назваются сукцинатами (лат. succinum – янтарь). Глутаровая кислота впервые была получена из глутаминовой аминокислоты, а та получила свое название от лат. gluten – клей, поскольку была найдена в клейковине пшеницы. Адипиновая кислота образуется при окислении жиров и получила название от лат. adeps – жир, сало. Эту кислоту синтезируют в промышленных масштабах, так как она является исходным веществом для производства полиамидных волокон (найлон-6,6) и смол. Кстати, название этого полимера происходит от первых букв двух городов – New York, London и числа атомов углерода в остатках адипиновой кислоты и гексаметилендиамина h3 N –(Ch3 )6 –Nh3, которые соединены поочередно в полимерную цепь. Название пимелиновой кислоты происходит от греч. pimelos – жир, субериновой (пробковой) кислоты – от лат. suber – пробка, себациновой кислоты – от лат. sebum – сало. Азелаиновая кислота была получена действием азотной кислоты на касторовое масло. Соответственно в ее названии можно найти «азо» и греч. elaion – масло.
Двухосновные кислоты с числом атомов углерода более 10 имеют обычно систематические названия. Но есть и исключения: брассиловая кислота была найдена в масле растений семейства Brassica; тапсиевая – в растении тапсия с греческого острова Тапсос, которое употреблялось в древности как лекарственное; японовая НООС–(СН2 )19 –СООН – выделена из высушенного сока некоторых акаций и пальм, растущих в Юго-Восточной Азии (раньше это вещество называли «японской землей»)
Основным способом получения сложных эфиров карбоновых кислот является реакция этерификации. Диэтилмалеат не является исключением. Этерификация – практически главнейший способ получения данного эфира двухосновной кислоты. Рассмотрим основные свойства реакции этерификации.
Итак, реакцией этерификации называется взаимодействие спиртов с карбоновыми кислотами, приводящее к образованию сложных эфиров:
В этой реакции молекула спирта выступает в роли нуклеофильного агента, атакующего бедный электронами углеродный атом карбонильной группы.
Реакции этерификации обратимы и, следовательно, ограничены состоянием равновесия. Превращение эквимолекулярных количеств кислоты и спирта в теоретически вычисленное количество сложного эфира по причине обратимости реакции невозможно. В результате реакции образуется некоторое максимальное количество эфира (которое всегда ниже теоретического) и остаются непрореагировавшие спирт и кислота. Например, при нагревании с обратным холодильником эквимолекулярных количеств уксусной кислоты и этилового спирта в реакцию вступает лишь 2/3 г-мол каждого компонента, поэтому максимальный выход эфира в этих условиях составляет лишь 2/3 теоретического, т. е. 66,7%.
По мере того как кислота и спирт реагируют друг с другом и происходит накопление продуктов их взаимодействия (эфира и воды), скорость обратной реакции, вначале незначительная, возрастает. При этом скорость прямой реакции постепенно уменьшается. Наконец, наступает динамическое равновесие, когда в единицу времени в сложный эфир превращается столько же молекул кислоты и спирта, сколько молекул сложного эфира распадается на кислоту и спирт. Одинаковой скоростью этих противоположно протекающих процессов обусловлен постоянный состав системы. Поскольку скорость бимолекулярной реакции пропорциональна произведению концентраций реагирующих веществ, мы можем для скоростей прямой и обратной реакций написать уравнения:
где v 1 — скорость реакции этерификации; v 2 — скорость реакции гидролиза; К1 и К2 — константы скорости обеих реакций; Ск, Сс, Сэ и Св — концентрации реагирующих и получающихся веществ (кислоты, спирта, эфира, воды).
В состоянии равновесия скорости реакций, протекающих в противоположных направлениях, равны, т. е. V 1 = V 2. Тогда К1 Ск Сс = Кг Сэ Св или:
Частное К2 /К1 является константой равновесия и обозначается буквой К.
Из полученного уравнения следует, что в состоянии равновесия отношение произведений концентраций реагирующих веществ обратно отношению констант скоростей реакций. В случае реакции образования уксусноэтилового эфира в состоянии равновесия, как упомянуто выше, в реакционной смеси содержится по 1/3 моля кислоты и спирта и по 2/3 моля эфира и воды. Поэтому
Однако можно изменить состояние равновесия и повысить выход сложного эфира, увеличивая концентрацию спирта (или кислоты). Например, если взять уксусную кислоту и спирт в молярном отношении, равном 1:2, выход эфира (из расчета на кислоту) повышается до 85%. Действительно, пусть концентрация эфира в состоянии равновесия (в молях) будет равна х, т. е. Сэ = х. Тогда и Св = х. Концентрация кислоты Ск = 1—х, концентрация спирта Сс = 2 — х. Следовательно,
После решения этого уравнения находим, что х = 0,85 моля, то есть выход эфира равен 85% теоретического.
Часто применяется и другой способ смещения равновесия в сторону большего выхода сложного эфира — удаление сложного эфира или воды из сферы реакции. Легко можно видеть, что уменьшение концентраций эфира или воды влечет уменьшение концентраций спирта и кислоты, поскольку величина константы равновесия К при данной температуре неизменна. Так, в случае получения низкокипящих сложных эфиров (например, уксусно-этилового с температурой кипения 77°С) в ходе реакции отгоняют эфир из реакционной колбы. При получении высококипящих сложных эфиров (например, уксуснобутилового с температурой кипения 125°С или уксусноизоамилового с температурой кипения 142°С) удобнее отгонять воду в процессе реакции. Вода в этом случае отгоняется в виде азеотропа с парами соответствующего спирта. При конденсации паров в холодильнике происходит расслоение этих ограниченно смешивающихся жидкостей и вода, как более тяжелая, собирается на дне поставленной на пути конденсата «ловушки» (см. рис. 27). Азеотропную отгонку воды можно использовать и в случае этерификации кислот этиловым или пропиловым спиртом, которые в жидкой фазе смешиваются с водой во всех отношениях. В этом случае для отделения воды от сконденсировавшегося в холодильнике спирта в реакционную смесь приходится добавлять третий компонент, образующий с водой и спиртом нераздельно кипящую смесь, но в жидкой фазе с водой не смешивающийся. Его роль состоит в том, что он экстрагирует из конденсата спирт и возвращает его в реакционный сосуд. В качестве такого компонента могут использоваться бензол, хлороформ, четыреххлористый углерод и некоторые другие жидкости, но из перечисленных только бензол можно использовать в «ловушках». Хлороформ и четыреххлористый углерод обладают большей плотностью, чем вода, и для отделения воды от реакционной смеси в случае использования этих жидкостей требуется «ловушка» другой конструкции.
При комнатной температуре реакция протекает очень медленно. При смешении эквимолярных количеств спирта и кислоты для достижения равновесных концентраций требуется до 16 лет. Повышение температуры ускоряет реакцию (так, в случае взаимодействия этилового спирта с уксусной кислотой при 110°С равновесие достигается через 10 дней, а при (155°С — через несколько часов).
Особенно сильное ускорение реакции этерификации достигается применением катализаторов — водородных ионов, получающихся при диссоциации сильных минеральных кислот. В качестве катализаторов чаще всего используются концентрированная серная кислота или сухой хлористый водород, ток которого пропускается через реакционную смесь. Найдено, что скорость реакции возрастает с увеличением количества катализатора; однако известно также, что добавка 0,01% серной кислоты достаточна для образования этилацетата из спирта и уксусной кислоты. Следует иметь в виду, что катализаторы повышают скорость реакции этерификации, но не могут вызывать сдвига равновесия.
Карбоновые кислоты, как видно из вышесказанного, реагируют со спиртами относительно медленно. Это объясняется слабой активностью карбонильной группы в кислотах по отношению к нуклеофильным агентам по сравнению с активностью той же группы в ангидридах и хлорангидридах кислот, поскольку +М -эффект гидроксильной группы приводит к уменьшению положительного заряда карбонильного углерода
Скорость этерификации карбоновой кислоты тем выше, чем больше положительный заряд карбонильного углерода. Величина δ+ на углероде карбоксильной группы зависит от характера радикала кислоты. Электронодонорные группы, связанные с карбоксилом, понижают дробный положительный заряд (по сравнению с зарядом в муравьиной кислоте) и тем препятствуют взаимодействию кислоты с нуклеофилом; электроноакцепторные заместители, напротив, делают кислоту более реакционноспособной. Поэтому кислоты типа трихлоруксусной, щавелевой, муравьиной быстро реагируют со спиртами даже без добавок минеральной кислоты-катализатора, а ароматические кислоты, особенно те, которые в ароматическом ядре содержат электронодонорные заместители, взаимодействуют со спиртом значительно труднее и требуют больших количеств катализатора.
Сильное влияние на скорость реакции этерификации оказывают также пространственные факторы. С увеличением объема связанных с карбоксилом углеводородных радикалов и с повышением объема этерифицируемых спиртов скорость этерификации уменьшается. Среди спиртов одного молекулярного веса быстрее всего взаимодействуют с кислотами первичные, медленнее — третичные спирты.
Реакцию этерификации можно проводить и в паровой фазе над твердыми катализаторами. Пары спирта и кислоты при 280—300° С пропускают через трубку с катализатором (ThO2 или TiO2 ). Выходы сложных эфиров в этом случае такие же, как и при реакциях в гомогенной фазе.
Аминокислоты образуют сложные эфиры при взаимодействии со спиртами в присутствии сухого хлористого водорода. Роль хлористого водорода здесь не ограничивается катализом реакции или сдвигом равновесия за счет связывания воды. В присутствии хлористого водорода аминокислота, находившаяся ранее в форме внутренней соли, превращается в хлористоводородную соль аминокислоты, причем карбоксильная группа из неактивной формы аниона переходит в реакционноспособную форму —СООН:
В результате этерификации в этих условиях эфиры также получаются в виде солей. Например, из аминоуксусной кислоты (гликоколя) и абсолютного этилового спирта образуется хлористоводородная соль эфира гликоколя
Свободный эфир из соли можно получить, удаляя хлористый водород окисью серебра:
Роль катализатора заключается в протонировании карбонильного кислорода: при этом карбонильный атом углерода становится более положительным и более «уязвимым» по отношению к атаке нуклеофильного агента, которым является молекула спирта. Образующийся вначале катион (VIII) присоединяет молекулу спирта за счет неподеленных электронов кислородного атома, давая катион (IX):
Далее катион (IX) отщепляет молекулу воды, превращаясь в катион сложного эфира (X):
Катион (X) в результате отщепления протона образует молекулу сложного эфира:
Использование метода «меченых атомов» дало возможность решить вопрос о месте разрыва связей при реакции этерификации. Оказалось, что обычно молекула воды образуется из гидроксила кислоты и водорода спирта. Следовательно, в молекуле кислоты разрывается связь между ацилом и гидроксилом, а в молекуле спирта — связь водорода с кислородом. Такой именно вывод следует из результатов работы по этерификации бензойной кислоты метанолом, содержащим тяжелый изотоп кислорода О18. Полученный сложный эфир содержал в своем составе указанный изотоп кислорода:
Присутствие О18 установлено сжиганием образца эфира и анализом образующихся продуктов сгорания (CO2 и Н2 О) на присутствие тяжелого изотопа кислорода.
Гидролиз сложных эфиров представляет собой реакцию, обратную реакции их образования. Гидролиз может быть осуществлен как в кислой, так и в щелочной среде. Для кислого гидролиза сложных эфиров справедливо все, что было сказано выше применительно к реакции этерификации, об обратимости и механизме процесса, о методах смещения равновесия. Щелочной гидролиз сложных эфиров проходит через следующие стадии:
Он является процессом необратимым, поскольку богатый электронами анион кислоты не способен взаимодействовать с нуклеофильной молекулой спирта.
Практически щелочной гидролиз сложных эфиров проводят в присутствии едких щелочей КОН, NaOH, а также гидроокисей щелочноземельных металлов Ва(ОН)2, Са(ОН)2 Образующиеся при гидролизе кислоты связываются в виде солей соответствующих металлов, поэтому гидроокиси приходится брать по крайней мере в эквивалентном отношении со сложным эфиром. Обычно используют избыток основания. Выделение кислот из их солей осуществляется с помощью сильных минеральных кислот.
В качестве растворителя основания для реакции гидролиза чаще всего применяют воду, которая, однако, не растворяет сложный эфир. Реакция идет на поверхности раздела двух фаз и требует поэтому хорошего перемешивания. Иногда реакцию бывает целесообразно проводить в гомогенной среде, используя в качестве растворителя водный спирт. При этом, однако, нужно иметь в виду, что для выделения кислоты перед подкислением раствора спирт необходимо удалить (отогнать).
1. Общая органическая химия. Карбоновые кислоты и их производные. Том 4. М., Химия, 1983, 729с.
2. Шабаров Ю.С. Органическая химия: В 2-х кн. — М.: Химия, 1994.- 848 с.
3. Петров А.А., Бальян Х.В., Трощенко А.Т. Органическая химия. – М.: Высш. шк., 1973. — 623 с.
4. Препаративная органическая химия. Изд. 2-е, М., Госхимиздат, 1964.
5. Богословский Б.Н., Казакова З.С. Скелетные катализаторы, их свойства и применение в органической химии. М., Госхимиздат, 1957.
6. Голодников Г.В. Практические работы по органическому синтезу. Л., Изд-во ЛГУ, 1966, 697с.
7. Дорофеенко Г.Н., Жданов Ю.А., Дуленко В.И. и др. Хлорная кислота и ее соединения ворганическом синтезе. Ростов, изд-во Ростовского ун-та, 1965.
8. Терней А. Современная органическая химия: В 2 т. — М.: Мир, 1981. — Т.1 — 670 с; Т.2 — 615 с.
9. Лабораторные работы по органической химии. Изд. 3-е. М., Высшая школа, 1974.
10. Храмкина М.Н. Практикум по органическому синтезу. Изд. 4-ое, Л., Химия. 1977.
11. В. Ф. Травень. Органическая химия. Том 1. – М.: Академкнига, 2004, — 708 с.
12. Голодников Г.В., Низовкина Т.В., Рыскальчук А.Т. Практикум по органическому синтезу. Л., Изд-во ЛГУ, 1967.
13. Крешков А.П., Курбатов И.Н. Лабораторные работы по синтезу и анализу органических соеднений. М., изд-во Артиллерийского ордена Ленина академии Красной армии им. Дзержинского, 1940.
www.ronl.ru
Статья - Синтез диэтилового эфира малоновой кислоты Свойства и основные методы получения сложных эфиров
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования
Самарский государственный технический университет
Кафедра: «Органическая химия»
“СИНТЕЗ ДИЭТИЛОВОГО ЭФИРА МАЛЕИНОВОЙ КИСЛОТЫ”
Курсовая работа
Выполнил:
Руководитель:
Самара, 2007 г.
Содержание
1. Введение
1.1. Свойства диэтилового эфира малеиновой кислоты
1.2. Практическое применение
1.3. Методика синтеза
2. Литературный обзор
2.1. Двухосновные (дикарбоновые) кислоты
2.2. Реакция этерификации
2.3. Механизм этерификации
3. Выводы
Список литературы
1. Введение
Свойства диэтилового эфира малеиновой кислоты
Диэтиловый эфир малеиновой кислоты, диэтилмалеат, этилмалеат C2H5OOCCH=CHCOOC2H5. Температура замерзания: ~-11.5°C, температура кипения: 225.3°C, дипольный момент: 2.54 Дебай, диэлектрическая проницаемость: 8.58 при 230С, плотность: 1.0687 при 20°С, г/мл, показатель преломления: 1.4400 при 20°С. Легковоспламеняющаяся жидкость
Практическое применение
Основная область применения диэтилмалеата – использование в качестве органического растворителя.
Органические растворители используются как в аналитической химии, так и в производстве. Среди потребителей органических растворителей – лаборатории и научно-исследовательские организации, предприятия нефтехимической, фармацевтической, парфюмерной, пищевой, электронной и оборонной промышленности.
Кроме того, широкое применение органические растворители, в частности диэтилмалеат, находят в лакокрасочной промышленности.
Постоянное ужесточение законодательства по охране окружающей среды привело к значительному вытеснению в последние годы традиционных красок на органических растворителях более экологически чистыми — водорастворимыми красками. Однако органоразбавляемые краски довольно часто используются в строительстве благодаря высокому качеству покрытий и относительному удобству применения. По разным оценкам их доля в общем объеме потребления строительных красок стабилизировалась на уровне 20-30%.
В настоящее время органоразбавляемые краски включены в программы большинства ведущих производителей лакокрасочных материалов. Чаще всего в качестве растворителя в современных органоразбавляемых красках применяют относительно низкотоксичный уайт-спирит, хотя иногда применяют и токсичные растворители (например, сольвент и ксилол). Кроме токсичности следствием применения в составе красок органических растворителей является их горючесть, а также характерный, часто достаточно сильный запах.
С появлением водоразбавляемых красок принято считать, что краски на органических растворителях имеют по сравнению ними всего два неоспоримых преимущества. Первое преимущество — возможность применения при отрицательных температурах (по материалам некоторых производителей до -20 0С). Второе преимущество — состоит в том, что свеженанесенное, еще не стабилизированное покрытие не может быть повреждено дождем.
Оба эти преимущества позволяют существенно расширить сезонность поведения работ, продлив ее на весну и осень. Теоретически возможно применение таких красок и в зимний период, однако это связано с рядом технологических сложностей, связанных с необходимостью предварительного оттаивания и осушения подложки.
Достаточно мощный импульс к использованию органоразбавляемых красок дало применения в качестве пленкообразователя специальный термопластиковой акриловой смолы Плиолит (PLIOLIT — торговая марка The Goodyear Tire & Rubber Co, USA).
Естественно краски на плиолитовых смолах обладают всеми перечисленными выше преимуществами органоразбавляемых красок, что однако не является главным. Главное же состоит в том, что они образуют достаточно хорошее покрытие, сравнимое с теми, которые можно получить с применением водоразбавляемых красок самых последних поколений.
На Российском рынке краски на основе плиолитовых смол представлены следующими фирмами: Alpa (Франция), Marshall (группа Akzo Nobel, Турция), Murolite (Швеция), Soframap (Франция)
Методика синтеза
/>
Малеиновая кислота 29 г (0,25 г-моль)
Этиловый спирт 96%-ный 32 г
Бензол 20 мл
Серная кислота (d=1,84). Бикарбонат натрия
/>
Приборы для проведения синтезов с азеотропной отгонкой воды: а– с холодильником Либиха, б– с шариковым холодильником, 1– реакционная колба, 2– двурогий форштосс, 3– капельная воронка, 4– «ловушка» для воды, 5– обратный холодильник.
В круглодонной колбе, снабженной обратным холодильником и ловушкой для воды, смешивают 29 г малеиновой кислоты, 32 г этилового спирта, 1,5 мл концентрированной серной кислоты и 20 мл бензола. Смесь кипятят на водяной бане или колбонагревателе до прекращения выделения воды, охлаждают, переносят в делительную воронку и промывают водой, последовательно водным раствором бикарбоната натрия и еще раз водой. После этого отгоняют растворитель, который захватывает с собой и следы воды. Остаток перегоняют из колбы с дефлегматором.
Выход диэтилмалеата 34 г (79% теоретического), температура кипения 123°Спри 12 мм рт. ст., />.
Литературный обзор.
Двухосновные (дикарбоновые) кислоты
Диэтилмалеат является сложным эфиром двухосновной малеиновой кислоты. Чтобы иметь представление о свойствах и структуре данного эфира, рассмотрим кратко этот класс органических соединений.
Общая формула этих кислот НООС–(СН2)n–СООН. Тривиальные названия имеют только первые члены ряда:
№
Название кислоты
Температура плавления
Растворимость, г/100 г Н2О при 20° С
Щавелевая
179,5
8,0
1
Малоновая
135,6
73,5
2
Янтарная
188 ,0
5,8
3
Глутаровая
97,5
63,9
4
Адипиновая
153,0
1,6
5
Пимелиновая
105,
5,0
6
Пробковая
144,0
0,16
7
Азелаиновая
106,5
0,24
8
Себациновая
134,5
0,1
9
Нонандикарбоновая
111
10
Декандикарбоновая
128
11
Брассиловая
113
12
Додекандикарбоновая
126
13
Тридекандикарбоновая
113,5
14
Тапсиевая
125
--PAGE_BREAK--
Зависимость температуры плавления от числа атомов углерода в молекулах представляет собой «пилу» с еще более острыми зубцами. Объяснение такое же, как и для монокарбоновых кислот: разное строение кристаллической решетки для четных и нечетных членов ряда. Для первых семи членов ряда наблюдается также сильное альтернирование величин растворимости кислот в воде. Понятно, что это свойство также связано с кристаллической решеткой: чем она прочнее, тем меньше растворимость.
Простейшая двухосновная щавелевая кислота содержит две соединенные карбоксильные группы НООС–СООН. Ее соли и эфиры называются оксалатами (от греч. oxys – кислый). Эта кислота известна с 17 в., она содержится (в виде калиевой соли) в щавеле (ее там 0,36%), откуда и получила свое название. Есть она и в других овощах и плодах: в шпинате ее 0,32%, в томатах – 0,06%. Избыток щавелевой кислоты может нарушать обмен веществ в организме, способствуя отложению нерастворимого оксалата кальция. Поэтому врачи рекомендуют ограничить потребление продуктов с повышенным содержанием этой кислоты.
Щавелевая кислота – одна из самых сильных органических кислот: при диссоциации по первой ступени она значительно сильнее уксусной. Она образует хорошо растворимые комплексные соединения со многими металлами, что используют для очистки металлов от ржавчины, для выведения ржавых пятен с тканей, сантехнических изделий и т.д. Например, ржавое пятно на белой ткани, смоченное раствором щавелевой кислоты, исчезает прямо на глазах.
Диэтиловый эфир малоновой кислоты (от лат. malum – яблоко) широко применяется в органических ситезах; химики называют его просто «малоновым эфиром». От этого же корня происходят названия непредельной малеиновой кислоты (цис-НООС–СН=СН–СООН) и производных яблочной кислоты – малатов. Интересно название транс-изомера малеиновой кислоты – фумаровой (от лат. fumus – дым). Эта кислота была обнаружена в растении Fumaria officinalis (дымянка), которое в античные времена сжигали, чтобы дымом отогнать злых духов.
Янтарная кислота была получена еще в 17 в. перегонкой янтаря, ее соли и эфиры назваются сукцинатами (лат. succinum – янтарь). Глутаровая кислота впервые была получена из глутаминовой аминокислоты, а та получила свое название от лат. gluten – клей, поскольку была найдена в клейковине пшеницы. Адипиновая кислота образуется при окислении жиров и получила название от лат. adeps – жир, сало. Эту кислоту синтезируют в промышленных масштабах, так как она является исходным веществом для производства полиамидных волокон (найлон-6,6) и смол. Кстати, название этого полимера происходит от первых букв двух городов – New York, London и числа атомов углерода в остатках адипиновой кислоты и гексаметилендиамина h3N –(Ch3)6 –Nh3, которые соединены поочередно в полимерную цепь. Название пимелиновой кислоты происходит от греч. pimelos – жир, субериновой (пробковой) кислоты – от лат. suber – пробка, себациновой кислоты – от лат. sebum – сало. Азелаиновая кислота была получена действием азотной кислоты на касторовое масло. Соответственно в ее названии можно найти «азо» и греч. elaion – масло.
Двухосновные кислоты с числом атомов углерода более 10 имеют обычно систематические названия. Но есть и исключения: брассиловая кислота была найдена в масле растений семейства Brassica; тапсиевая – в растении тапсия с греческого острова Тапсос, которое употреблялось в древности как лекарственное; японовая НООС–(СН2)19–СООН – выделена из высушенного сока некоторых акаций и пальм, растущих в Юго-Восточной Азии (раньше это вещество называли «японской землей»)
Реакция этерификации
Основным способом получения сложных эфиров карбоновых кислот является реакция этерификации. Диэтилмалеат не является исключением. Этерификация – практически главнейший способ получения данного эфира двухосновной кислоты. Рассмотрим основные свойства реакции этерификации.
Итак, реакцией этерификации называется взаимодействие спиртов с карбоновыми кислотами, приводящее к образованию сложных эфиров:
/>
В этой реакции молекула спирта выступает в роли нуклеофильного агента, атакующего бедный электронами углеродный атом карбонильной группы.
Реакции этерификации обратимы и, следовательно, ограничены состоянием равновесия. Превращение эквимолекулярных количеств кислоты и спирта в теоретически вычисленное количество сложного эфира по причине обратимости реакции невозможно. В результате реакции образуется некоторое максимальное количество эфира (которое всегда ниже теоретического) и остаются непрореагировавшие спирт и кислота. Например, при нагревании с обратным холодильником эквимолекулярных количеств уксусной кислоты и этилового спирта в реакцию вступает лишь 2/3 г-мол каждого компонента, поэтому максимальный выход эфира в этих условиях составляет лишь 2/3 теоретического, т. е. 66,7%.
/>
По мере того как кислота и спирт реагируют друг с другом и происходит накопление продуктов их взаимодействия (эфира и воды), скорость обратной реакции, вначале незначительная, возрастает. При этом скорость прямой реакции постепенно уменьшается. Наконец, наступает динамическое равновесие, когда в единицу времени в сложный эфир превращается столько же молекул кислоты и спирта, сколько молекул сложного эфира распадается на кислоту и спирт. Одинаковой скоростью этих противоположно протекающих процессов обусловлен постоянный состав системы. Поскольку скорость бимолекулярной реакции пропорциональна произведению концентраций реагирующих веществ, мы можем для скоростей прямой и обратной реакций написать уравнения:
/>
где v1— скорость реакции этерификации; v2— скорость реакции гидролиза; К1и К2— константы скорости обеих реакций; Ск, Сс, Сэи Св— концентрации реагирующих и получающихся веществ (кислоты, спирта, эфира, воды).
В состоянии равновесия скорости реакций, протекающих в противоположных направлениях, равны, т. е. V1= V2. Тогда К1СкСс= КгСэСвили:
/>
Частное К2/К1является константой равновесия и обозначается буквой К.
Из полученного уравнения следует, что в состоянии равновесия отношение произведений концентраций реагирующих веществ обратно отношению констант скоростей реакций. В случае реакции образования уксусноэтилового эфира в состоянии равновесия, как упомянуто выше, в реакционной смеси содержится по 1/3 моля кислоты и спирта и по 2/3 моля эфира и воды. Поэтому
/>
Однако можно изменить состояние равновесия и повысить выход сложного эфира, увеличивая концентрацию спирта (или кислоты). Например, если взять уксусную кислоту и спирт в молярном отношении, равном 1:2, выход эфира (из расчета на кислоту) повышается до 85%. Действительно, пусть концентрация эфира в состоянии равновесия (в молях) будет равна х, т. е. Сэ= х. Тогда и Св= х. Концентрация кислоты Ск= 1—х, концентрация спирта Сс= 2 — х. Следовательно,
/>
После решения этого уравнения находим, что х = 0,85 моля, то есть выход эфира равен 85% теоретического.
Часто применяется и другой способ смещения равновесия в сторону большего выхода сложного эфира — удаление сложного эфира или воды из сферы реакции. Легко можно видеть, что уменьшение концентраций эфира или воды влечет уменьшение концентраций спирта и кислоты, поскольку величина константы равновесия К при данной температуре неизменна. Так, в случае получения низкокипящих сложных эфиров (например, уксусно-этилового с температурой кипения 77°С) в ходе реакции отгоняют эфир из реакционной колбы. При получении высококипящих сложных эфиров (например, уксуснобутилового с температурой кипения 125°С или уксусноизоамилового с температурой кипения 142°С) удобнее отгонять воду в процессе реакции. Вода в этом случае отгоняется в виде азеотропа с парами соответствующего спирта. При конденсации паров в холодильнике происходит расслоение этих ограниченно смешивающихся жидкостей и вода, как более тяжелая, собирается на дне поставленной на пути конденсата «ловушки» (см. рис. 27). Азеотропную отгонку воды можно использовать и в случае этерификации кислот этиловым или пропиловым спиртом, которые в жидкой фазе смешиваются с водой во всех отношениях. В этом случае для отделения воды от сконденсировавшегося в холодильнике спирта в реакционную смесь приходится добавлять третий компонент, образующий с водой и спиртом нераздельно кипящую смесь, но в жидкой фазе с водой не смешивающийся. Его роль состоит в том, что он экстрагирует из конденсата спирт и возвращает его в реакционный сосуд. В качестве такого компонента могут использоваться бензол, хлороформ, четыреххлористый углерод и некоторые другие жидкости, но из перечисленных только бензол можно использовать в «ловушках». Хлороформ и четыреххлористый углерод обладают большей плотностью, чем вода, и для отделения воды от реакционной смеси в случае использования этих жидкостей требуется «ловушка» другой конструкции.
продолжение --PAGE_BREAK--При комнатной температуре реакция протекает очень медленно. При смешении эквимолярных количеств спирта и кислоты для достижения равновесных концентраций требуется до 16 лет. Повышение температуры ускоряет реакцию (так, в случае взаимодействия этилового спирта с уксусной кислотой при 110°Сравновесие достигается через 10 дней, а при (155°С— через несколько часов).
Особенно сильное ускорение реакции этерификации достигается применением катализаторов — водородных ионов, получающихся при диссоциации сильных минеральных кислот. В качестве катализаторов чаще всего используются концентрированная серная кислота или сухой хлористый водород, ток которого пропускается через реакционную смесь. Найдено, что скорость реакции возрастает с увеличением количества катализатора; однако известно также, что добавка 0,01% серной кислоты достаточна для образования этилацетата из спирта и уксусной кислоты. Следует иметь в виду, что катализаторы повышают скорость реакции этерификации, но не могут вызывать сдвига равновесия.
Карбоновые кислоты, как видно из вышесказанного, реагируют со спиртами относительно медленно. Это объясняется слабой активностью карбонильной группы в кислотах по отношению к нуклеофильным агентам по сравнению с активностью той же группы в ангидридах и хлорангидридах кислот, поскольку +М-эффект гидроксильной группы приводит к уменьшению положительного заряда карбонильного углерода
/>
Скорость этерификации карбоновой кислоты тем выше, чем больше положительный заряд карбонильного углерода. Величина δ+ на углероде карбоксильной группы зависит от характера радикала кислоты. Электронодонорные группы, связанные с карбоксилом, понижают дробный положительный заряд (по сравнению с зарядом в муравьиной кислоте) и тем препятствуют взаимодействию кислоты с нуклеофилом; электроноакцепторные заместители, напротив, делают кислоту более реакционноспособной. Поэтому кислоты типа трихлоруксусной, щавелевой, муравьиной быстро реагируют со спиртами даже без добавок минеральной кислоты-катализатора, а ароматические кислоты, особенно те, которые в ароматическом ядре содержат электронодонорные заместители, взаимодействуют со спиртом значительно труднее и требуют больших количеств катализатора.
Сильное влияние на скорость реакции этерификации оказывают также пространственные факторы. С увеличением объема связанных с карбоксилом углеводородных радикалов и с повышением объема этерифицируемых спиртов скорость этерификации уменьшается. Среди спиртов одного молекулярного веса быстрее всего взаимодействуют с кислотами первичные, медленнее — третичные спирты.
Реакцию этерификации можно проводить и в паровой фазе над твердыми катализаторами. Пары спирта и кислоты при 280—300° С пропускают через трубку с катализатором (ThO2 или TiO2). Выходы сложных эфиров в этом случае такие же, как и при реакциях в гомогенной фазе.
Аминокислоты образуют сложные эфиры при взаимодействии со спиртами в присутствии сухого хлористого водорода. Роль хлористого водорода здесь не ограничивается катализом реакции или сдвигом равновесия за счет связывания воды. В присутствии хлористого водорода аминокислота, находившаяся ранее в форме внутренней соли, превращается в хлористоводородную соль аминокислоты, причем карбоксильная группа из неактивной формы аниона переходит в реакционноспособную форму —СООН:
/>
В результате этерификации в этих условиях эфиры также получаются в виде солей. Например, из аминоуксусной кислоты (гликоколя) и абсолютного этилового спирта образуется хлористоводородная соль эфира гликоколя
/>
Свободный эфир из соли можно получить, удаляя хлористый водород окисью серебра:
/>
Механизм этерификации
Роль катализатора заключается в протонировании карбонильного кислорода: при этом карбонильный атом углерода становится более положительным и более «уязвимым» по отношению к атаке нуклеофильного агента, которым является молекула спирта. Образующийся вначале катион (VIII) присоединяет молекулу спирта за счет неподеленных электронов кислородного атома, давая катион (IX):
/>
Далее катион (IX) отщепляет молекулу воды, превращаясь в катион сложного эфира (X):
/>
Катион (X) в результате отщепления протона образует молекулу сложного эфира:
/>
Использование метода «меченых атомов» дало возможность решить вопрос о месте разрыва связей при реакции этерификации. Оказалось, что обычно молекула воды образуется из гидроксила кислоты и водорода спирта. Следовательно, в молекуле кислоты разрывается связь между ацилом и гидроксилом, а в молекуле спирта — связь водорода с кислородом. Такой именно вывод следует из результатов работы по этерификации бензойной кислоты метанолом, содержащим тяжелый изотоп кислорода О18. Полученный сложный эфир содержал в своем составе указанный изотоп кислорода:
Присутствие О18установлено сжиганием образца эфира и анализом образующихся продуктов сгорания (CO2и Н2О) на присутствие тяжелого изотопа кислорода.
Гидролиз сложных эфиров представляет собой реакцию, обратную реакции их образования. Гидролиз может быть осуществлен как в кислой, так и в щелочной среде. Для кислого гидролиза сложных эфиров справедливо все, что было сказано выше применительно к реакции этерификации, об обратимости и механизме процесса, о методах смещения равновесия. Щелочной гидролиз сложных эфиров проходит через следующие стадии:
/>
Он является процессом необратимым, поскольку богатый электронами анион кислоты не способен взаимодействовать с нуклеофильной молекулой спирта.
Практически щелочной гидролиз сложных эфиров проводят в присутствии едких щелочей КОН, NaOH, а также гидроокисей щелочноземельных металлов Ва(ОН)2, Са(ОН)2Образующиеся при гидролизе кислоты связываются в виде солей соответствующих металлов, поэтому гидроокиси приходится брать по крайней мере в эквивалентном отношении со сложным эфиром. Обычно используют избыток основания. Выделение кислот из их солей осуществляется с помощью сильных минеральных кислот.
В качестве растворителя основания для реакции гидролиза чаще всего применяют воду, которая, однако, не растворяет сложный эфир. Реакция идет на поверхности раздела двух фаз и требует поэтому хорошего перемешивания. Иногда реакцию бывает целесообразно проводить в гомогенной среде, используя в качестве растворителя водный спирт. При этом, однако, нужно иметь в виду, что для выделения кислоты перед подкислением раствора спирт необходимо удалить (отогнать).
Список литературы
Общая органическая химия. Карбоновые кислоты и их производные. Том 4. М., Химия, 1983, 729с.
Шабаров Ю.С. Органическая химия: В 2-х кн. — М.: Химия, 1994.- 848 с.
Петров А.А., Бальян Х.В., Трощенко А.Т. Органическая химия. – М.: Высш. шк., 1973. — 623 с.
Препаративная органическая химия. Изд. 2-е, М., Госхимиздат, 1964.
Богословский Б.Н., Казакова З.С. Скелетные катализаторы, их свойства и применение в органической химии. М., Госхимиздат, 1957.
Голодников Г.В. Практические работы по органическому синтезу. Л., Изд-во ЛГУ, 1966, 697с.
Дорофеенко Г.Н., Жданов Ю.А., Дуленко В.И. и др. Хлорная кислота и ее соединения ворганическом синтезе. Ростов, изд-во Ростовского ун-та, 1965.
Терней А. Современная органическая химия: В 2 т. — М.: Мир, 1981. — Т.1 — 670 с; Т.2 — 615 с.
Лабораторные работы по органической химии. Изд. 3-е. М., Высшая школа, 1974.
Храмкина М.Н. Практикум по органическому синтезу. Изд. 4-ое, Л., Химия. 1977.
В. Ф. Травень. Органическая химия. Том 1. – М.: Академкнига, 2004, — 708 с.
Голодников Г.В., Низовкина Т.В., Рыскальчук А.Т. Практикум по органическому синтезу. Л., Изд-во ЛГУ, 1967.
Крешков А.П., Курбатов И.Н. Лабораторные работы по синтезу и анализу органических соеднений. М., изд-во Артиллерийского ордена Ленина академии Красной армии им. Дзержинского, 1940.
www.ronl.ru
Лекция - Синтез диэтилового эфира малоновой кислоты Свойства и основные методы получения сложных эфиров
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования
Самарский государственный технический университет
Кафедра: «Органическая химия»
“СИНТЕЗ ДИЭТИЛОВОГО ЭФИРА МАЛЕИНОВОЙ КИСЛОТЫ”
Курсовая работа
Выполнил:
Руководитель:
Самара, 2007 г.
Содержание
1. Введение
1.1. Свойства диэтилового эфира малеиновой кислоты
1.2. Практическое применение
1.3. Методика синтеза
2. Литературный обзор
2.1. Двухосновные (дикарбоновые) кислоты
2.2. Реакция этерификации
2.3. Механизм этерификации
3. Выводы
Список литературы
1. Введение
Свойства диэтилового эфира малеиновой кислоты
Диэтиловый эфир малеиновой кислоты, диэтилмалеат, этилмалеат C2H5OOCCH=CHCOOC2H5. Температура замерзания: ~-11.5°C, температура кипения: 225.3°C, дипольный момент: 2.54 Дебай, диэлектрическая проницаемость: 8.58 при 230С, плотность: 1.0687 при 20°С, г/мл, показатель преломления: 1.4400 при 20°С. Легковоспламеняющаяся жидкость
Практическое применение
Основная область применения диэтилмалеата – использование в качестве органического растворителя.
Органические растворители используются как в аналитической химии, так и в производстве. Среди потребителей органических растворителей – лаборатории и научно-исследовательские организации, предприятия нефтехимической, фармацевтической, парфюмерной, пищевой, электронной и оборонной промышленности.
Кроме того, широкое применение органические растворители, в частности диэтилмалеат, находят в лакокрасочной промышленности.
Постоянное ужесточение законодательства по охране окружающей среды привело к значительному вытеснению в последние годы традиционных красок на органических растворителях более экологически чистыми — водорастворимыми красками. Однако органоразбавляемые краски довольно часто используются в строительстве благодаря высокому качеству покрытий и относительному удобству применения. По разным оценкам их доля в общем объеме потребления строительных красок стабилизировалась на уровне 20-30%.
В настоящее время органоразбавляемые краски включены в программы большинства ведущих производителей лакокрасочных материалов. Чаще всего в качестве растворителя в современных органоразбавляемых красках применяют относительно низкотоксичный уайт-спирит, хотя иногда применяют и токсичные растворители (например, сольвент и ксилол). Кроме токсичности следствием применения в составе красок органических растворителей является их горючесть, а также характерный, часто достаточно сильный запах.
С появлением водоразбавляемых красок принято считать, что краски на органических растворителях имеют по сравнению ними всего два неоспоримых преимущества. Первое преимущество — возможность применения при отрицательных температурах (по материалам некоторых производителей до -20 0С). Второе преимущество — состоит в том, что свеженанесенное, еще не стабилизированное покрытие не может быть повреждено дождем.
Оба эти преимущества позволяют существенно расширить сезонность поведения работ, продлив ее на весну и осень. Теоретически возможно применение таких красок и в зимний период, однако это связано с рядом технологических сложностей, связанных с необходимостью предварительного оттаивания и осушения подложки.
Достаточно мощный импульс к использованию органоразбавляемых красок дало применения в качестве пленкообразователя специальный термопластиковой акриловой смолы Плиолит (PLIOLIT — торговая марка The Goodyear Tire & Rubber Co, USA).
Естественно краски на плиолитовых смолах обладают всеми перечисленными выше преимуществами органоразбавляемых красок, что однако не является главным. Главное же состоит в том, что они образуют достаточно хорошее покрытие, сравнимое с теми, которые можно получить с применением водоразбавляемых красок самых последних поколений.
На Российском рынке краски на основе плиолитовых смол представлены следующими фирмами: Alpa (Франция), Marshall (группа Akzo Nobel, Турция), Murolite (Швеция), Soframap (Франция)
Методика синтеза
/>
Малеиновая кислота 29 г (0,25 г-моль)
Этиловый спирт 96%-ный 32 г
Бензол 20 мл
Серная кислота (d=1,84). Бикарбонат натрия
/>
Приборы для проведения синтезов с азеотропной отгонкой воды: а– с холодильником Либиха, б– с шариковым холодильником, 1– реакционная колба, 2– двурогий форштосс, 3– капельная воронка, 4– «ловушка» для воды, 5– обратный холодильник.
В круглодонной колбе, снабженной обратным холодильником и ловушкой для воды, смешивают 29 г малеиновой кислоты, 32 г этилового спирта, 1,5 мл концентрированной серной кислоты и 20 мл бензола. Смесь кипятят на водяной бане или колбонагревателе до прекращения выделения воды, охлаждают, переносят в делительную воронку и промывают водой, последовательно водным раствором бикарбоната натрия и еще раз водой. После этого отгоняют растворитель, который захватывает с собой и следы воды. Остаток перегоняют из колбы с дефлегматором.
Выход диэтилмалеата 34 г (79% теоретического), температура кипения 123°Спри 12 мм рт. ст., />.
Литературный обзор.
Двухосновные (дикарбоновые) кислоты
Диэтилмалеат является сложным эфиром двухосновной малеиновой кислоты. Чтобы иметь представление о свойствах и структуре данного эфира, рассмотрим кратко этот класс органических соединений.
Общая формула этих кислот НООС–(СН2)n–СООН. Тривиальные названия имеют только первые члены ряда:
№
Название кислоты
Температура плавления
Растворимость, г/100 г Н2О при 20° С
Щавелевая
179,5
8,0
1
Малоновая
135,6
73,5
2
Янтарная
188 ,0
5,8
3
Глутаровая
97,5
63,9
4
Адипиновая
153,0
1,6
5
Пимелиновая
105,
5,0
6
Пробковая
144,0
0,16
7
Азелаиновая
106,5
0,24
8
Себациновая
134,5
0,1
9
Нонандикарбоновая
111
10
Декандикарбоновая
128
11
Брассиловая
113
12
Додекандикарбоновая
126
13
Тридекандикарбоновая
113,5
14
Тапсиевая
125
--PAGE_BREAK--
Зависимость температуры плавления от числа атомов углерода в молекулах представляет собой «пилу» с еще более острыми зубцами. Объяснение такое же, как и для монокарбоновых кислот: разное строение кристаллической решетки для четных и нечетных членов ряда. Для первых семи членов ряда наблюдается также сильное альтернирование величин растворимости кислот в воде. Понятно, что это свойство также связано с кристаллической решеткой: чем она прочнее, тем меньше растворимость.
Простейшая двухосновная щавелевая кислота содержит две соединенные карбоксильные группы НООС–СООН. Ее соли и эфиры называются оксалатами (от греч. oxys – кислый). Эта кислота известна с 17 в., она содержится (в виде калиевой соли) в щавеле (ее там 0,36%), откуда и получила свое название. Есть она и в других овощах и плодах: в шпинате ее 0,32%, в томатах – 0,06%. Избыток щавелевой кислоты может нарушать обмен веществ в организме, способствуя отложению нерастворимого оксалата кальция. Поэтому врачи рекомендуют ограничить потребление продуктов с повышенным содержанием этой кислоты.
Щавелевая кислота – одна из самых сильных органических кислот: при диссоциации по первой ступени она значительно сильнее уксусной. Она образует хорошо растворимые комплексные соединения со многими металлами, что используют для очистки металлов от ржавчины, для выведения ржавых пятен с тканей, сантехнических изделий и т.д. Например, ржавое пятно на белой ткани, смоченное раствором щавелевой кислоты, исчезает прямо на глазах.
Диэтиловый эфир малоновой кислоты (от лат. malum – яблоко) широко применяется в органических ситезах; химики называют его просто «малоновым эфиром». От этого же корня происходят названия непредельной малеиновой кислоты (цис-НООС–СН=СН–СООН) и производных яблочной кислоты – малатов. Интересно название транс-изомера малеиновой кислоты – фумаровой (от лат. fumus – дым). Эта кислота была обнаружена в растении Fumaria officinalis (дымянка), которое в античные времена сжигали, чтобы дымом отогнать злых духов.
Янтарная кислота была получена еще в 17 в. перегонкой янтаря, ее соли и эфиры назваются сукцинатами (лат. succinum – янтарь). Глутаровая кислота впервые была получена из глутаминовой аминокислоты, а та получила свое название от лат. gluten – клей, поскольку была найдена в клейковине пшеницы. Адипиновая кислота образуется при окислении жиров и получила название от лат. adeps – жир, сало. Эту кислоту синтезируют в промышленных масштабах, так как она является исходным веществом для производства полиамидных волокон (найлон-6,6) и смол. Кстати, название этого полимера происходит от первых букв двух городов – New York, London и числа атомов углерода в остатках адипиновой кислоты и гексаметилендиамина h3N –(Ch3)6 –Nh3, которые соединены поочередно в полимерную цепь. Название пимелиновой кислоты происходит от греч. pimelos – жир, субериновой (пробковой) кислоты – от лат. suber – пробка, себациновой кислоты – от лат. sebum – сало. Азелаиновая кислота была получена действием азотной кислоты на касторовое масло. Соответственно в ее названии можно найти «азо» и греч. elaion – масло.
Двухосновные кислоты с числом атомов углерода более 10 имеют обычно систематические названия. Но есть и исключения: брассиловая кислота была найдена в масле растений семейства Brassica; тапсиевая – в растении тапсия с греческого острова Тапсос, которое употреблялось в древности как лекарственное; японовая НООС–(СН2)19–СООН – выделена из высушенного сока некоторых акаций и пальм, растущих в Юго-Восточной Азии (раньше это вещество называли «японской землей»)
Реакция этерификации
Основным способом получения сложных эфиров карбоновых кислот является реакция этерификации. Диэтилмалеат не является исключением. Этерификация – практически главнейший способ получения данного эфира двухосновной кислоты. Рассмотрим основные свойства реакции этерификации.
Итак, реакцией этерификации называется взаимодействие спиртов с карбоновыми кислотами, приводящее к образованию сложных эфиров:
/>
В этой реакции молекула спирта выступает в роли нуклеофильного агента, атакующего бедный электронами углеродный атом карбонильной группы.
Реакции этерификации обратимы и, следовательно, ограничены состоянием равновесия. Превращение эквимолекулярных количеств кислоты и спирта в теоретически вычисленное количество сложного эфира по причине обратимости реакции невозможно. В результате реакции образуется некоторое максимальное количество эфира (которое всегда ниже теоретического) и остаются непрореагировавшие спирт и кислота. Например, при нагревании с обратным холодильником эквимолекулярных количеств уксусной кислоты и этилового спирта в реакцию вступает лишь 2/3 г-мол каждого компонента, поэтому максимальный выход эфира в этих условиях составляет лишь 2/3 теоретического, т. е. 66,7%.
/>
По мере того как кислота и спирт реагируют друг с другом и происходит накопление продуктов их взаимодействия (эфира и воды), скорость обратной реакции, вначале незначительная, возрастает. При этом скорость прямой реакции постепенно уменьшается. Наконец, наступает динамическое равновесие, когда в единицу времени в сложный эфир превращается столько же молекул кислоты и спирта, сколько молекул сложного эфира распадается на кислоту и спирт. Одинаковой скоростью этих противоположно протекающих процессов обусловлен постоянный состав системы. Поскольку скорость бимолекулярной реакции пропорциональна произведению концентраций реагирующих веществ, мы можем для скоростей прямой и обратной реакций написать уравнения:
/>
где v1— скорость реакции этерификации; v2— скорость реакции гидролиза; К1и К2— константы скорости обеих реакций; Ск, Сс, Сэи Св— концентрации реагирующих и получающихся веществ (кислоты, спирта, эфира, воды).
В состоянии равновесия скорости реакций, протекающих в противоположных направлениях, равны, т. е. V1= V2. Тогда К1СкСс= КгСэСвили:
/>
Частное К2/К1является константой равновесия и обозначается буквой К.
Из полученного уравнения следует, что в состоянии равновесия отношение произведений концентраций реагирующих веществ обратно отношению констант скоростей реакций. В случае реакции образования уксусноэтилового эфира в состоянии равновесия, как упомянуто выше, в реакционной смеси содержится по 1/3 моля кислоты и спирта и по 2/3 моля эфира и воды. Поэтому
/>
Однако можно изменить состояние равновесия и повысить выход сложного эфира, увеличивая концентрацию спирта (или кислоты). Например, если взять уксусную кислоту и спирт в молярном отношении, равном 1:2, выход эфира (из расчета на кислоту) повышается до 85%. Действительно, пусть концентрация эфира в состоянии равновесия (в молях) будет равна х, т. е. Сэ= х. Тогда и Св= х. Концентрация кислоты Ск= 1—х, концентрация спирта Сс= 2 — х. Следовательно,
/>
После решения этого уравнения находим, что х = 0,85 моля, то есть выход эфира равен 85% теоретического.
Часто применяется и другой способ смещения равновесия в сторону большего выхода сложного эфира — удаление сложного эфира или воды из сферы реакции. Легко можно видеть, что уменьшение концентраций эфира или воды влечет уменьшение концентраций спирта и кислоты, поскольку величина константы равновесия К при данной температуре неизменна. Так, в случае получения низкокипящих сложных эфиров (например, уксусно-этилового с температурой кипения 77°С) в ходе реакции отгоняют эфир из реакционной колбы. При получении высококипящих сложных эфиров (например, уксуснобутилового с температурой кипения 125°С или уксусноизоамилового с температурой кипения 142°С) удобнее отгонять воду в процессе реакции. Вода в этом случае отгоняется в виде азеотропа с парами соответствующего спирта. При конденсации паров в холодильнике происходит расслоение этих ограниченно смешивающихся жидкостей и вода, как более тяжелая, собирается на дне поставленной на пути конденсата «ловушки» (см. рис. 27). Азеотропную отгонку воды можно использовать и в случае этерификации кислот этиловым или пропиловым спиртом, которые в жидкой фазе смешиваются с водой во всех отношениях. В этом случае для отделения воды от сконденсировавшегося в холодильнике спирта в реакционную смесь приходится добавлять третий компонент, образующий с водой и спиртом нераздельно кипящую смесь, но в жидкой фазе с водой не смешивающийся. Его роль состоит в том, что он экстрагирует из конденсата спирт и возвращает его в реакционный сосуд. В качестве такого компонента могут использоваться бензол, хлороформ, четыреххлористый углерод и некоторые другие жидкости, но из перечисленных только бензол можно использовать в «ловушках». Хлороформ и четыреххлористый углерод обладают большей плотностью, чем вода, и для отделения воды от реакционной смеси в случае использования этих жидкостей требуется «ловушка» другой конструкции.
продолжение --PAGE_BREAK--При комнатной температуре реакция протекает очень медленно. При смешении эквимолярных количеств спирта и кислоты для достижения равновесных концентраций требуется до 16 лет. Повышение температуры ускоряет реакцию (так, в случае взаимодействия этилового спирта с уксусной кислотой при 110°Сравновесие достигается через 10 дней, а при (155°С— через несколько часов).
Особенно сильное ускорение реакции этерификации достигается применением катализаторов — водородных ионов, получающихся при диссоциации сильных минеральных кислот. В качестве катализаторов чаще всего используются концентрированная серная кислота или сухой хлористый водород, ток которого пропускается через реакционную смесь. Найдено, что скорость реакции возрастает с увеличением количества катализатора; однако известно также, что добавка 0,01% серной кислоты достаточна для образования этилацетата из спирта и уксусной кислоты. Следует иметь в виду, что катализаторы повышают скорость реакции этерификации, но не могут вызывать сдвига равновесия.
Карбоновые кислоты, как видно из вышесказанного, реагируют со спиртами относительно медленно. Это объясняется слабой активностью карбонильной группы в кислотах по отношению к нуклеофильным агентам по сравнению с активностью той же группы в ангидридах и хлорангидридах кислот, поскольку +М-эффект гидроксильной группы приводит к уменьшению положительного заряда карбонильного углерода
/>
Скорость этерификации карбоновой кислоты тем выше, чем больше положительный заряд карбонильного углерода. Величина δ+ на углероде карбоксильной группы зависит от характера радикала кислоты. Электронодонорные группы, связанные с карбоксилом, понижают дробный положительный заряд (по сравнению с зарядом в муравьиной кислоте) и тем препятствуют взаимодействию кислоты с нуклеофилом; электроноакцепторные заместители, напротив, делают кислоту более реакционноспособной. Поэтому кислоты типа трихлоруксусной, щавелевой, муравьиной быстро реагируют со спиртами даже без добавок минеральной кислоты-катализатора, а ароматические кислоты, особенно те, которые в ароматическом ядре содержат электронодонорные заместители, взаимодействуют со спиртом значительно труднее и требуют больших количеств катализатора.
Сильное влияние на скорость реакции этерификации оказывают также пространственные факторы. С увеличением объема связанных с карбоксилом углеводородных радикалов и с повышением объема этерифицируемых спиртов скорость этерификации уменьшается. Среди спиртов одного молекулярного веса быстрее всего взаимодействуют с кислотами первичные, медленнее — третичные спирты.
Реакцию этерификации можно проводить и в паровой фазе над твердыми катализаторами. Пары спирта и кислоты при 280—300° С пропускают через трубку с катализатором (ThO2 или TiO2). Выходы сложных эфиров в этом случае такие же, как и при реакциях в гомогенной фазе.
Аминокислоты образуют сложные эфиры при взаимодействии со спиртами в присутствии сухого хлористого водорода. Роль хлористого водорода здесь не ограничивается катализом реакции или сдвигом равновесия за счет связывания воды. В присутствии хлористого водорода аминокислота, находившаяся ранее в форме внутренней соли, превращается в хлористоводородную соль аминокислоты, причем карбоксильная группа из неактивной формы аниона переходит в реакционноспособную форму —СООН:
/>
В результате этерификации в этих условиях эфиры также получаются в виде солей. Например, из аминоуксусной кислоты (гликоколя) и абсолютного этилового спирта образуется хлористоводородная соль эфира гликоколя
/>
Свободный эфир из соли можно получить, удаляя хлористый водород окисью серебра:
/>
Механизм этерификации
Роль катализатора заключается в протонировании карбонильного кислорода: при этом карбонильный атом углерода становится более положительным и более «уязвимым» по отношению к атаке нуклеофильного агента, которым является молекула спирта. Образующийся вначале катион (VIII) присоединяет молекулу спирта за счет неподеленных электронов кислородного атома, давая катион (IX):
/>
Далее катион (IX) отщепляет молекулу воды, превращаясь в катион сложного эфира (X):
/>
Катион (X) в результате отщепления протона образует молекулу сложного эфира:
/>
Использование метода «меченых атомов» дало возможность решить вопрос о месте разрыва связей при реакции этерификации. Оказалось, что обычно молекула воды образуется из гидроксила кислоты и водорода спирта. Следовательно, в молекуле кислоты разрывается связь между ацилом и гидроксилом, а в молекуле спирта — связь водорода с кислородом. Такой именно вывод следует из результатов работы по этерификации бензойной кислоты метанолом, содержащим тяжелый изотоп кислорода О18. Полученный сложный эфир содержал в своем составе указанный изотоп кислорода:
Присутствие О18установлено сжиганием образца эфира и анализом образующихся продуктов сгорания (CO2и Н2О) на присутствие тяжелого изотопа кислорода.
Гидролиз сложных эфиров представляет собой реакцию, обратную реакции их образования. Гидролиз может быть осуществлен как в кислой, так и в щелочной среде. Для кислого гидролиза сложных эфиров справедливо все, что было сказано выше применительно к реакции этерификации, об обратимости и механизме процесса, о методах смещения равновесия. Щелочной гидролиз сложных эфиров проходит через следующие стадии:
/>
Он является процессом необратимым, поскольку богатый электронами анион кислоты не способен взаимодействовать с нуклеофильной молекулой спирта.
Практически щелочной гидролиз сложных эфиров проводят в присутствии едких щелочей КОН, NaOH, а также гидроокисей щелочноземельных металлов Ва(ОН)2, Са(ОН)2Образующиеся при гидролизе кислоты связываются в виде солей соответствующих металлов, поэтому гидроокиси приходится брать по крайней мере в эквивалентном отношении со сложным эфиром. Обычно используют избыток основания. Выделение кислот из их солей осуществляется с помощью сильных минеральных кислот.
В качестве растворителя основания для реакции гидролиза чаще всего применяют воду, которая, однако, не растворяет сложный эфир. Реакция идет на поверхности раздела двух фаз и требует поэтому хорошего перемешивания. Иногда реакцию бывает целесообразно проводить в гомогенной среде, используя в качестве растворителя водный спирт. При этом, однако, нужно иметь в виду, что для выделения кислоты перед подкислением раствора спирт необходимо удалить (отогнать).
Список литературы
Общая органическая химия. Карбоновые кислоты и их производные. Том 4. М., Химия, 1983, 729с.
Шабаров Ю.С. Органическая химия: В 2-х кн. — М.: Химия, 1994.- 848 с.
Петров А.А., Бальян Х.В., Трощенко А.Т. Органическая химия. – М.: Высш. шк., 1973. — 623 с.
Препаративная органическая химия. Изд. 2-е, М., Госхимиздат, 1964.
Богословский Б.Н., Казакова З.С. Скелетные катализаторы, их свойства и применение в органической химии. М., Госхимиздат, 1957.
Голодников Г.В. Практические работы по органическому синтезу. Л., Изд-во ЛГУ, 1966, 697с.
Дорофеенко Г.Н., Жданов Ю.А., Дуленко В.И. и др. Хлорная кислота и ее соединения ворганическом синтезе. Ростов, изд-во Ростовского ун-та, 1965.
Терней А. Современная органическая химия: В 2 т. — М.: Мир, 1981. — Т.1 — 670 с; Т.2 — 615 с.
Лабораторные работы по органической химии. Изд. 3-е. М., Высшая школа, 1974.
Храмкина М.Н. Практикум по органическому синтезу. Изд. 4-ое, Л., Химия. 1977.
В. Ф. Травень. Органическая химия. Том 1. – М.: Академкнига, 2004, — 708 с.
Голодников Г.В., Низовкина Т.В., Рыскальчук А.Т. Практикум по органическому синтезу. Л., Изд-во ЛГУ, 1967.
Крешков А.П., Курбатов И.Н. Лабораторные работы по синтезу и анализу органических соеднений. М., изд-во Артиллерийского ордена Ленина академии Красной армии им. Дзержинского, 1940.
www.ronl.ru
Статья - Синтез диэтилового эфира малоновой кислоты. Свойства и основные методы получения сложных эфиров
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования
Самарский государственный технический университет
Кафедра: «Органическая химия»
“СИНТЕЗ ДИЭТИЛОВОГО ЭФИРА МАЛЕИНОВОЙ КИСЛОТЫ”
Курсовая работа
Выполнил:
Руководитель:
Самара, 2007 г.
Содержание
1. Введение
1.1. Свойства диэтилового эфира малеиновой кислоты
1.2. Практическое применение
1.3. Методика синтеза
2. Литературный обзор
2.1. Двухосновные (дикарбоновые) кислоты
2.2. Реакция этерификации
2.3. Механизм этерификации
3. Выводы
Список литературы
1. Введение
1.1. Свойства диэтилового эфира малеиновой кислоты
Диэтиловый эфир малеиновой кислоты, диэтилмалеат, этилмалеат C2 H5 OOCCH=CHCOOC2 H5. Температура замерзания: ~-11.5°C, температура кипения: 225.3°C, дипольный момент: 2.54 Дебай, диэлектрическая проницаемость: 8.58 при 230С, плотность: 1.0687 при 20°С, г/мл, показатель преломления: 1.4400 при 20°С. Легковоспламеняющаяся жидкость
Основная область применения диэтилмалеата – использование в качестве органического растворителя.
Органические растворители используются как в аналитической химии, так и в производстве. Среди потребителей органических растворителей – лаборатории и научно-исследовательские организации, предприятия нефтехимической, фармацевтической, парфюмерной, пищевой, электронной и оборонной промышленности.
Кроме того, широкое применение органические растворители, в частности диэтилмалеат, находят в лакокрасочной промышленности.
Постоянное ужесточение законодательства по охране окружающей среды привело к значительному вытеснению в последние годы традиционных красок на органических растворителях более экологически чистыми — водорастворимыми красками. Однако органоразбавляемые краски довольно часто используются в строительстве благодаря высокому качеству покрытий и относительному удобству применения. По разным оценкам их доля в общем объеме потребления строительных красок стабилизировалась на уровне 20-30%.
В настоящее время органоразбавляемые краски включены в программы большинства ведущих производителей лакокрасочных материалов. Чаще всего в качестве растворителя в современных органоразбавляемых красках применяют относительно низкотоксичный уайт-спирит, хотя иногда применяют и токсичные растворители (например, сольвент и ксилол). Кроме токсичности следствием применения в составе красок органических растворителей является их горючесть, а также характерный, часто достаточно сильный запах.
С появлением водоразбавляемых красок принято считать, что краски на органических растворителях имеют по сравнению ними всего два неоспоримых преимущества. Первое преимущество — возможность применения при отрицательных температурах (по материалам некоторых производителей до -20 0С). Второе преимущество — состоит в том, что свеженанесенное, еще не стабилизированное покрытие не может быть повреждено дождем.
Оба эти преимущества позволяют существенно расширить сезонность поведения работ, продлив ее на весну и осень. Теоретически возможно применение таких красок и в зимний период, однако это связано с рядом технологических сложностей, связанных с необходимостью предварительного оттаивания и осушения подложки.
Достаточно мощный импульс к использованию органоразбавляемых красок дало применения в качестве пленкообразователя специальный термопластиковой акриловой смолы Плиолит (PLIOLIT — торговая марка The Goodyear Tire & Rubber Co, USA).
Естественно краски на плиолитовых смолах обладают всеми перечисленными выше преимуществами органоразбавляемых красок, что однако не является главным. Главное же состоит в том, что они образуют достаточно хорошее покрытие, сравнимое с теми, которые можно получить с применением водоразбавляемых красок самых последних поколений.
На Российском рынке краски на основе плиолитовых смол представлены следующими фирмами: Alpa (Франция), Marshall (группа Akzo Nobel, Турция), Murolite (Швеция), Soframap (Франция)
Малеиновая кислота… 29 г (0,25 г-моль)
Этиловый спирт 96%-ный… 32 г
Бензол… 20 мл
Серная кислота ( d = 1,84). Бикарбонат натрия
Приборы для проведения синтезов с азеотропной отгонкой воды: а – с холодильником Либиха, б – с шариковым холодильником, 1 – реакционная колба, 2 – двурогий форштосс, 3 – капельная воронка, 4 – «ловушка» для воды, 5 – обратный холодильник.
В круглодонной колбе, снабженной обратным холодильником и ловушкой для воды, смешивают 29 г малеиновой кислоты, 32 г этилового спирта, 1,5 мл концентрированной серной кислоты и 20 мл бензола. Смесь кипятят на водяной бане или колбонагревателе до прекращения выделения воды, охлаждают, переносят в делительную воронку и промывают водой, последовательно водным раствором бикарбоната натрия и еще раз водой. После этого отгоняют растворитель, который захватывает с собой и следы воды. Остаток перегоняют из колбы с дефлегматором.
Выход диэтилмалеата 34 г (79% теоретического), температура кипения 123°С при 12 мм рт. ст., .
Диэтилмалеат является сложным эфиром двухосновной малеиновой кислоты. Чтобы иметь представление о свойствах и структуре данного эфира, рассмотрим кратко этот класс органических соединений.
Общая формула этих кислот НООС–(СН2 )n –СООН. Тривиальные названия имеют только первые члены ряда:
№ | Название кислоты | Температура плавления | Растворимость, г/100 г Н2 О при 20° С |
Щавелевая | 179,5 | 8,0 | |
1 | Малоновая | 135,6 | 73,5 |
2 | Янтарная | 188 ,0 | 5,8 |
3 | Глутаровая | 97,5 | 63,9 |
4 | Адипиновая | 153,0 | 1,6 |
5 | Пимелиновая | 105, | 5,0 |
6 | Пробковая | 144,0 | 0,16 |
7 | Азелаиновая | 106,5 | 0,24 |
8 | Себациновая | 134,5 | 0,1 |
9 | Нонандикарбоновая | 111 | |
10 | Декандикарбоновая | 128 | |
11 | Брассиловая | 113 | |
12 | Додекандикарбоновая | 126 | |
13 | Тридекандикарбоновая | 113,5 | |
14 | Тапсиевая | 125 |
Зависимость температуры плавления от числа атомов углерода в молекулах представляет собой «пилу» с еще более острыми зубцами. Объяснение такое же, как и для монокарбоновых кислот: разное строение кристаллической решетки для четных и нечетных членов ряда. Для первых семи членов ряда наблюдается также сильное альтернирование величин растворимости кислот в воде. Понятно, что это свойство также связано с кристаллической решеткой: чем она прочнее, тем меньше растворимость.
Простейшая двухосновная щавелевая кислота содержит две соединенные карбоксильные группы НООС–СООН. Ее соли и эфиры называются оксалатами (от греч. oxys – кислый). Эта кислота известна с 17 в., она содержится (в виде калиевой соли) в щавеле (ее там 0,36%), откуда и получила свое название. Есть она и в других овощах и плодах: в шпинате ее 0,32%, в томатах – 0,06%. Избыток щавелевой кислоты может нарушать обмен веществ в организме, способствуя отложению нерастворимого оксалата кальция. Поэтому врачи рекомендуют ограничить потребление продуктов с повышенным содержанием этой кислоты.
Щавелевая кислота – одна из самых сильных органических кислот: при диссоциации по первой ступени она значительно сильнее уксусной. Она образует хорошо растворимые комплексные соединения со многими металлами, что используют для очистки металлов от ржавчины, для выведения ржавых пятен с тканей, сантехнических изделий и т.д. Например, ржавое пятно на белой ткани, смоченное раствором щавелевой кислоты, исчезает прямо на глазах.
Диэтиловый эфир малоновой кислоты (от лат. malum – яблоко) широко применяется в органических ситезах; химики называют его просто «малоновым эфиром». От этого же корня происходят названия непредельной малеиновой кислоты (цис -НООС–СН=СН–СООН) и производных яблочной кислоты – малатов. Интересно название транс -изомера малеиновой кислоты – фумаровой (от лат. fumus – дым). Эта кислота была обнаружена в растении Fumaria officinalis (дымянка), которое в античные времена сжигали, чтобы дымом отогнать злых духов.
Янтарная кислота была получена еще в 17 в. перегонкой янтаря, ее соли и эфиры назваются сукцинатами (лат. succinum – янтарь). Глутаровая кислота впервые была получена из глутаминовой аминокислоты, а та получила свое название от лат. gluten – клей, поскольку была найдена в клейковине пшеницы. Адипиновая кислота образуется при окислении жиров и получила название от лат. adeps – жир, сало. Эту кислоту синтезируют в промышленных масштабах, так как она является исходным веществом для производства полиамидных волокон (найлон-6,6) и смол. Кстати, название этого полимера происходит от первых букв двух городов – New York, London и числа атомов углерода в остатках адипиновой кислоты и гексаметилендиамина h3 N –(Ch3 )6 –Nh3, которые соединены поочередно в полимерную цепь. Название пимелиновой кислоты происходит от греч. pimelos – жир, субериновой (пробковой) кислоты – от лат. suber – пробка, себациновой кислоты – от лат. sebum – сало. Азелаиновая кислота была получена действием азотной кислоты на касторовое масло. Соответственно в ее названии можно найти «азо» и греч. elaion – масло.
Двухосновные кислоты с числом атомов углерода более 10 имеют обычно систематические названия. Но есть и исключения: брассиловая кислота была найдена в масле растений семейства Brassica; тапсиевая – в растении тапсия с греческого острова Тапсос, которое употреблялось в древности как лекарственное; японовая НООС–(СН2 )19 –СООН – выделена из высушенного сока некоторых акаций и пальм, растущих в Юго-Восточной Азии (раньше это вещество называли «японской землей»)
Основным способом получения сложных эфиров карбоновых кислот является реакция этерификации. Диэтилмалеат не является исключением. Этерификация – практически главнейший способ получения данного эфира двухосновной кислоты. Рассмотрим основные свойства реакции этерификации.
Итак, реакцией этерификации называется взаимодействие спиртов с карбоновыми кислотами, приводящее к образованию сложных эфиров:
В этой реакции молекула спирта выступает в роли нуклеофильного агента, атакующего бедный электронами углеродный атом карбонильной группы.
Реакции этерификации обратимы и, следовательно, ограничены состоянием равновесия. Превращение эквимолекулярных количеств кислоты и спирта в теоретически вычисленное количество сложного эфира по причине обратимости реакции невозможно. В результате реакции образуется некоторое максимальное количество эфира (которое всегда ниже теоретического) и остаются непрореагировавшие спирт и кислота. Например, при нагревании с обратным холодильником эквимолекулярных количеств уксусной кислоты и этилового спирта в реакцию вступает лишь 2/3 г-мол каждого компонента, поэтому максимальный выход эфира в этих условиях составляет лишь 2/3 теоретического, т. е. 66,7%.
По мере того как кислота и спирт реагируют друг с другом и происходит накопление продуктов их взаимодействия (эфира и воды), скорость обратной реакции, вначале незначительная, возрастает. При этом скорость прямой реакции постепенно уменьшается. Наконец, наступает динамическое равновесие, когда в единицу времени в сложный эфир превращается столько же молекул кислоты и спирта, сколько молекул сложного эфира распадается на кислоту и спирт. Одинаковой скоростью этих противоположно протекающих процессов обусловлен постоянный состав системы. Поскольку скорость бимолекулярной реакции пропорциональна произведению концентраций реагирующих веществ, мы можем для скоростей прямой и обратной реакций написать уравнения:
где v 1 — скорость реакции этерификации; v 2 — скорость реакции гидролиза; К1 и К2 — константы скорости обеих реакций; Ск, Сс, Сэ и Св — концентрации реагирующих и получающихся веществ (кислоты, спирта, эфира, воды).
В состоянии равновесия скорости реакций, протекающих в противоположных направлениях, равны, т. е. V 1 = V 2. Тогда К1 Ск Сс = Кг Сэ Св или:
Частное К2 /К1 является константой равновесия и обозначается буквой К.
Из полученного уравнения следует, что в состоянии равновесия отношение произведений концентраций реагирующих веществ обратно отношению констант скоростей реакций. В случае реакции образования уксусноэтилового эфира в состоянии равновесия, как упомянуто выше, в реакционной смеси содержится по 1/3 моля кислоты и спирта и по 2/3 моля эфира и воды. Поэтому
Однако можно изменить состояние равновесия и повысить выход сложного эфира, увеличивая концентрацию спирта (или кислоты). Например, если взять уксусную кислоту и спирт в молярном отношении, равном 1:2, выход эфира (из расчета на кислоту) повышается до 85%. Действительно, пусть концентрация эфира в состоянии равновесия (в молях) будет равна х, т. е. Сэ = х. Тогда и Св = х. Концентрация кислоты Ск = 1—х, концентрация спирта Сс = 2 — х. Следовательно,
После решения этого уравнения находим, что х = 0,85 моля, то есть выход эфира равен 85% теоретического.
Часто применяется и другой способ смещения равновесия в сторону большего выхода сложного эфира — удаление сложного эфира или воды из сферы реакции. Легко можно видеть, что уменьшение концентраций эфира или воды влечет уменьшение концентраций спирта и кислоты, поскольку величина константы равновесия К при данной температуре неизменна. Так, в случае получения низкокипящих сложных эфиров (например, уксусно-этилового с температурой кипения 77°С) в ходе реакции отгоняют эфир из реакционной колбы. При получении высококипящих сложных эфиров (например, уксуснобутилового с температурой кипения 125°С или уксусноизоамилового с температурой кипения 142°С) удобнее отгонять воду в процессе реакции. Вода в этом случае отгоняется в виде азеотропа с парами соответствующего спирта. При конденсации паров в холодильнике происходит расслоение этих ограниченно смешивающихся жидкостей и вода, как более тяжелая, собирается на дне поставленной на пути конденсата «ловушки» (см. рис. 27). Азеотропную отгонку воды можно использовать и в случае этерификации кислот этиловым или пропиловым спиртом, которые в жидкой фазе смешиваются с водой во всех отношениях. В этом случае для отделения воды от сконденсировавшегося в холодильнике спирта в реакционную смесь приходится добавлять третий компонент, образующий с водой и спиртом нераздельно кипящую смесь, но в жидкой фазе с водой не смешивающийся. Его роль состоит в том, что он экстрагирует из конденсата спирт и возвращает его в реакционный сосуд. В качестве такого компонента могут использоваться бензол, хлороформ, четыреххлористый углерод и некоторые другие жидкости, но из перечисленных только бензол можно использовать в «ловушках». Хлороформ и четыреххлористый углерод обладают большей плотностью, чем вода, и для отделения воды от реакционной смеси в случае использования этих жидкостей требуется «ловушка» другой конструкции.
При комнатной температуре реакция протекает очень медленно. При смешении эквимолярных количеств спирта и кислоты для достижения равновесных концентраций требуется до 16 лет. Повышение температуры ускоряет реакцию (так, в случае взаимодействия этилового спирта с уксусной кислотой при 110°С равновесие достигается через 10 дней, а при (155°С — через несколько часов).
Особенно сильное ускорение реакции этерификации достигается применением катализаторов — водородных ионов, получающихся при диссоциации сильных минеральных кислот. В качестве катализаторов чаще всего используются концентрированная серная кислота или сухой хлористый водород, ток которого пропускается через реакционную смесь. Найдено, что скорость реакции возрастает с увеличением количества катализатора; однако известно также, что добавка 0,01% серной кислоты достаточна для образования этилацетата из спирта и уксусной кислоты. Следует иметь в виду, что катализаторы повышают скорость реакции этерификации, но не могут вызывать сдвига равновесия.
Карбоновые кислоты, как видно из вышесказанного, реагируют со спиртами относительно медленно. Это объясняется слабой активностью карбонильной группы в кислотах по отношению к нуклеофильным агентам по сравнению с активностью той же группы в ангидридах и хлорангидридах кислот, поскольку +М -эффект гидроксильной группы приводит к уменьшению положительного заряда карбонильного углерода
Скорость этерификации карбоновой кислоты тем выше, чем больше положительный заряд карбонильного углерода. Величина δ+ на углероде карбоксильной группы зависит от характера радикала кислоты. Электронодонорные группы, связанные с карбоксилом, понижают дробный положительный заряд (по сравнению с зарядом в муравьиной кислоте) и тем препятствуют взаимодействию кислоты с нуклеофилом; электроноакцепторные заместители, напротив, делают кислоту более реакционноспособной. Поэтому кислоты типа трихлоруксусной, щавелевой, муравьиной быстро реагируют со спиртами даже без добавок минеральной кислоты-катализатора, а ароматические кислоты, особенно те, которые в ароматическом ядре содержат электронодонорные заместители, взаимодействуют со спиртом значительно труднее и требуют больших количеств катализатора.
Сильное влияние на скорость реакции этерификации оказывают также пространственные факторы. С увеличением объема связанных с карбоксилом углеводородных радикалов и с повышением объема этерифицируемых спиртов скорость этерификации уменьшается. Среди спиртов одного молекулярного веса быстрее всего взаимодействуют с кислотами первичные, медленнее — третичные спирты.
Реакцию этерификации можно проводить и в паровой фазе над твердыми катализаторами. Пары спирта и кислоты при 280—300° С пропускают через трубку с катализатором (ThO2 или TiO2 ). Выходы сложных эфиров в этом случае такие же, как и при реакциях в гомогенной фазе.
Аминокислоты образуют сложные эфиры при взаимодействии со спиртами в присутствии сухого хлористого водорода. Роль хлористого водорода здесь не ограничивается катализом реакции или сдвигом равновесия за счет связывания воды. В присутствии хлористого водорода аминокислота, находившаяся ранее в форме внутренней соли, превращается в хлористоводородную соль аминокислоты, причем карбоксильная группа из неактивной формы аниона переходит в реакционноспособную форму —СООН:
В результате этерификации в этих условиях эфиры также получаются в виде солей. Например, из аминоуксусной кислоты (гликоколя) и абсолютного этилового спирта образуется хлористоводородная соль эфира гликоколя
Свободный эфир из соли можно получить, удаляя хлористый водород окисью серебра:
Роль катализатора заключается в протонировании карбонильного кислорода: при этом карбонильный атом углерода становится более положительным и более «уязвимым» по отношению к атаке нуклеофильного агента, которым является молекула спирта. Образующийся вначале катион (VIII) присоединяет молекулу спирта за счет неподеленных электронов кислородного атома, давая катион (IX):
Далее катион (IX) отщепляет молекулу воды, превращаясь в катион сложного эфира (X):
Катион (X) в результате отщепления протона образует молекулу сложного эфира:
Использование метода «меченых атомов» дало возможность решить вопрос о месте разрыва связей при реакции этерификации. Оказалось, что обычно молекула воды образуется из гидроксила кислоты и водорода спирта. Следовательно, в молекуле кислоты разрывается связь между ацилом и гидроксилом, а в молекуле спирта — связь водорода с кислородом. Такой именно вывод следует из результатов работы по этерификации бензойной кислоты метанолом, содержащим тяжелый изотоп кислорода О18. Полученный сложный эфир содержал в своем составе указанный изотоп кислорода:
Присутствие О18 установлено сжиганием образца эфира и анализом образующихся продуктов сгорания (CO2 и Н2 О) на присутствие тяжелого изотопа кислорода.
Гидролиз сложных эфиров представляет собой реакцию, обратную реакции их образования. Гидролиз может быть осуществлен как в кислой, так и в щелочной среде. Для кислого гидролиза сложных эфиров справедливо все, что было сказано выше применительно к реакции этерификации, об обратимости и механизме процесса, о методах смещения равновесия. Щелочной гидролиз сложных эфиров проходит через следующие стадии:
Он является процессом необратимым, поскольку богатый электронами анион кислоты не способен взаимодействовать с нуклеофильной молекулой спирта.
Практически щелочной гидролиз сложных эфиров проводят в присутствии едких щелочей КОН, NaOH, а также гидроокисей щелочноземельных металлов Ва(ОН)2, Са(ОН)2 Образующиеся при гидролизе кислоты связываются в виде солей соответствующих металлов, поэтому гидроокиси приходится брать по крайней мере в эквивалентном отношении со сложным эфиром. Обычно используют избыток основания. Выделение кислот из их солей осуществляется с помощью сильных минеральных кислот.
В качестве растворителя основания для реакции гидролиза чаще всего применяют воду, которая, однако, не растворяет сложный эфир. Реакция идет на поверхности раздела двух фаз и требует поэтому хорошего перемешивания. Иногда реакцию бывает целесообразно проводить в гомогенной среде, используя в качестве растворителя водный спирт. При этом, однако, нужно иметь в виду, что для выделения кислоты перед подкислением раствора спирт необходимо удалить (отогнать).
1. Общая органическая химия. Карбоновые кислоты и их производные. Том 4. М., Химия, 1983, 729с.
2. Шабаров Ю.С. Органическая химия: В 2-х кн. — М.: Химия, 1994.- 848 с.
3. Петров А.А., Бальян Х.В., Трощенко А.Т. Органическая химия. – М.: Высш. шк., 1973. — 623 с.
4. Препаративная органическая химия. Изд. 2-е, М., Госхимиздат, 1964.
5. Богословский Б.Н., Казакова З.С. Скелетные катализаторы, их свойства и применение в органической химии. М., Госхимиздат, 1957.
6. Голодников Г.В. Практические работы по органическому синтезу. Л., Изд-во ЛГУ, 1966, 697с.
7. Дорофеенко Г.Н., Жданов Ю.А., Дуленко В.И. и др. Хлорная кислота и ее соединения ворганическом синтезе. Ростов, изд-во Ростовского ун-та, 1965.
8. Терней А. Современная органическая химия: В 2 т. — М.: Мир, 1981. — Т.1 — 670 с; Т.2 — 615 с.
9. Лабораторные работы по органической химии. Изд. 3-е. М., Высшая школа, 1974.
10. Храмкина М.Н. Практикум по органическому синтезу. Изд. 4-ое, Л., Химия. 1977.
11. В. Ф. Травень. Органическая химия. Том 1. – М.: Академкнига, 2004, — 708 с.
12. Голодников Г.В., Низовкина Т.В., Рыскальчук А.Т. Практикум по органическому синтезу. Л., Изд-во ЛГУ, 1967.
13. Крешков А.П., Курбатов И.Н. Лабораторные работы по синтезу и анализу органических соеднений. М., изд-во Артиллерийского ордена Ленина академии Красной армии им. Дзержинского, 1940.
www.ronl.ru
Методичка - Синтез диэтилового эфира малоновой кислоты Свойства и основные методы получения сложных эфиров
Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального образования
Самарский государственный технический университет
Кафедра: «Органическая химия»
“СИНТЕЗ ДИЭТИЛОВОГО ЭФИРА МАЛЕИНОВОЙ КИСЛОТЫ”
Курсовая работа
Выполнил:
Руководитель:
Самара, 2007 г.
Содержание
1. Введение
1.1. Свойства диэтилового эфира малеиновой кислоты
1.2. Практическое применение
1.3. Методика синтеза
2. Литературный обзор
2.1. Двухосновные (дикарбоновые) кислоты
2.2. Реакция этерификации
2.3. Механизм этерификации
3. Выводы
Список литературы
1. Введение
Свойства диэтилового эфира малеиновой кислоты
Диэтиловый эфир малеиновой кислоты, диэтилмалеат, этилмалеат C2H5OOCCH=CHCOOC2H5. Температура замерзания: ~-11.5°C, температура кипения: 225.3°C, дипольный момент: 2.54 Дебай, диэлектрическая проницаемость: 8.58 при 230С, плотность: 1.0687 при 20°С, г/мл, показатель преломления: 1.4400 при 20°С. Легковоспламеняющаяся жидкость
Практическое применение
Основная область применения диэтилмалеата – использование в качестве органического растворителя.
Органические растворители используются как в аналитической химии, так и в производстве. Среди потребителей органических растворителей – лаборатории и научно-исследовательские организации, предприятия нефтехимической, фармацевтической, парфюмерной, пищевой, электронной и оборонной промышленности.
Кроме того, широкое применение органические растворители, в частности диэтилмалеат, находят в лакокрасочной промышленности.
Постоянное ужесточение законодательства по охране окружающей среды привело к значительному вытеснению в последние годы традиционных красок на органических растворителях более экологически чистыми — водорастворимыми красками. Однако органоразбавляемые краски довольно часто используются в строительстве благодаря высокому качеству покрытий и относительному удобству применения. По разным оценкам их доля в общем объеме потребления строительных красок стабилизировалась на уровне 20-30%.
В настоящее время органоразбавляемые краски включены в программы большинства ведущих производителей лакокрасочных материалов. Чаще всего в качестве растворителя в современных органоразбавляемых красках применяют относительно низкотоксичный уайт-спирит, хотя иногда применяют и токсичные растворители (например, сольвент и ксилол). Кроме токсичности следствием применения в составе красок органических растворителей является их горючесть, а также характерный, часто достаточно сильный запах.
С появлением водоразбавляемых красок принято считать, что краски на органических растворителях имеют по сравнению ними всего два неоспоримых преимущества. Первое преимущество — возможность применения при отрицательных температурах (по материалам некоторых производителей до -20 0С). Второе преимущество — состоит в том, что свеженанесенное, еще не стабилизированное покрытие не может быть повреждено дождем.
Оба эти преимущества позволяют существенно расширить сезонность поведения работ, продлив ее на весну и осень. Теоретически возможно применение таких красок и в зимний период, однако это связано с рядом технологических сложностей, связанных с необходимостью предварительного оттаивания и осушения подложки.
Достаточно мощный импульс к использованию органоразбавляемых красок дало применения в качестве пленкообразователя специальный термопластиковой акриловой смолы Плиолит (PLIOLIT — торговая марка The Goodyear Tire & Rubber Co, USA).
Естественно краски на плиолитовых смолах обладают всеми перечисленными выше преимуществами органоразбавляемых красок, что однако не является главным. Главное же состоит в том, что они образуют достаточно хорошее покрытие, сравнимое с теми, которые можно получить с применением водоразбавляемых красок самых последних поколений.
На Российском рынке краски на основе плиолитовых смол представлены следующими фирмами: Alpa (Франция), Marshall (группа Akzo Nobel, Турция), Murolite (Швеция), Soframap (Франция)
Методика синтеза
/>
Малеиновая кислота 29 г (0,25 г-моль)
Этиловый спирт 96%-ный 32 г
Бензол 20 мл
Серная кислота (d=1,84). Бикарбонат натрия
/>
Приборы для проведения синтезов с азеотропной отгонкой воды: а– с холодильником Либиха, б– с шариковым холодильником, 1– реакционная колба, 2– двурогий форштосс, 3– капельная воронка, 4– «ловушка» для воды, 5– обратный холодильник.
В круглодонной колбе, снабженной обратным холодильником и ловушкой для воды, смешивают 29 г малеиновой кислоты, 32 г этилового спирта, 1,5 мл концентрированной серной кислоты и 20 мл бензола. Смесь кипятят на водяной бане или колбонагревателе до прекращения выделения воды, охлаждают, переносят в делительную воронку и промывают водой, последовательно водным раствором бикарбоната натрия и еще раз водой. После этого отгоняют растворитель, который захватывает с собой и следы воды. Остаток перегоняют из колбы с дефлегматором.
Выход диэтилмалеата 34 г (79% теоретического), температура кипения 123°Спри 12 мм рт. ст., />.
Литературный обзор.
Двухосновные (дикарбоновые) кислоты
Диэтилмалеат является сложным эфиром двухосновной малеиновой кислоты. Чтобы иметь представление о свойствах и структуре данного эфира, рассмотрим кратко этот класс органических соединений.
Общая формула этих кислот НООС–(СН2)n–СООН. Тривиальные названия имеют только первые члены ряда:
№
Название кислоты
Температура плавления
Растворимость, г/100 г Н2О при 20° С
Щавелевая
179,5
8,0
1
Малоновая
135,6
73,5
2
Янтарная
188 ,0
5,8
3
Глутаровая
97,5
63,9
4
Адипиновая
153,0
1,6
5
Пимелиновая
105,
5,0
6
Пробковая
144,0
0,16
7
Азелаиновая
106,5
0,24
8
Себациновая
134,5
0,1
9
Нонандикарбоновая
111
10
Декандикарбоновая
128
11
Брассиловая
113
12
Додекандикарбоновая
126
13
Тридекандикарбоновая
113,5
14
Тапсиевая
125
--PAGE_BREAK--
Зависимость температуры плавления от числа атомов углерода в молекулах представляет собой «пилу» с еще более острыми зубцами. Объяснение такое же, как и для монокарбоновых кислот: разное строение кристаллической решетки для четных и нечетных членов ряда. Для первых семи членов ряда наблюдается также сильное альтернирование величин растворимости кислот в воде. Понятно, что это свойство также связано с кристаллической решеткой: чем она прочнее, тем меньше растворимость.
Простейшая двухосновная щавелевая кислота содержит две соединенные карбоксильные группы НООС–СООН. Ее соли и эфиры называются оксалатами (от греч. oxys – кислый). Эта кислота известна с 17 в., она содержится (в виде калиевой соли) в щавеле (ее там 0,36%), откуда и получила свое название. Есть она и в других овощах и плодах: в шпинате ее 0,32%, в томатах – 0,06%. Избыток щавелевой кислоты может нарушать обмен веществ в организме, способствуя отложению нерастворимого оксалата кальция. Поэтому врачи рекомендуют ограничить потребление продуктов с повышенным содержанием этой кислоты.
Щавелевая кислота – одна из самых сильных органических кислот: при диссоциации по первой ступени она значительно сильнее уксусной. Она образует хорошо растворимые комплексные соединения со многими металлами, что используют для очистки металлов от ржавчины, для выведения ржавых пятен с тканей, сантехнических изделий и т.д. Например, ржавое пятно на белой ткани, смоченное раствором щавелевой кислоты, исчезает прямо на глазах.
Диэтиловый эфир малоновой кислоты (от лат. malum – яблоко) широко применяется в органических ситезах; химики называют его просто «малоновым эфиром». От этого же корня происходят названия непредельной малеиновой кислоты (цис-НООС–СН=СН–СООН) и производных яблочной кислоты – малатов. Интересно название транс-изомера малеиновой кислоты – фумаровой (от лат. fumus – дым). Эта кислота была обнаружена в растении Fumaria officinalis (дымянка), которое в античные времена сжигали, чтобы дымом отогнать злых духов.
Янтарная кислота была получена еще в 17 в. перегонкой янтаря, ее соли и эфиры назваются сукцинатами (лат. succinum – янтарь). Глутаровая кислота впервые была получена из глутаминовой аминокислоты, а та получила свое название от лат. gluten – клей, поскольку была найдена в клейковине пшеницы. Адипиновая кислота образуется при окислении жиров и получила название от лат. adeps – жир, сало. Эту кислоту синтезируют в промышленных масштабах, так как она является исходным веществом для производства полиамидных волокон (найлон-6,6) и смол. Кстати, название этого полимера происходит от первых букв двух городов – New York, London и числа атомов углерода в остатках адипиновой кислоты и гексаметилендиамина h3N –(Ch3)6 –Nh3, которые соединены поочередно в полимерную цепь. Название пимелиновой кислоты происходит от греч. pimelos – жир, субериновой (пробковой) кислоты – от лат. suber – пробка, себациновой кислоты – от лат. sebum – сало. Азелаиновая кислота была получена действием азотной кислоты на касторовое масло. Соответственно в ее названии можно найти «азо» и греч. elaion – масло.
Двухосновные кислоты с числом атомов углерода более 10 имеют обычно систематические названия. Но есть и исключения: брассиловая кислота была найдена в масле растений семейства Brassica; тапсиевая – в растении тапсия с греческого острова Тапсос, которое употреблялось в древности как лекарственное; японовая НООС–(СН2)19–СООН – выделена из высушенного сока некоторых акаций и пальм, растущих в Юго-Восточной Азии (раньше это вещество называли «японской землей»)
Реакция этерификации
Основным способом получения сложных эфиров карбоновых кислот является реакция этерификации. Диэтилмалеат не является исключением. Этерификация – практически главнейший способ получения данного эфира двухосновной кислоты. Рассмотрим основные свойства реакции этерификации.
Итак, реакцией этерификации называется взаимодействие спиртов с карбоновыми кислотами, приводящее к образованию сложных эфиров:
/>
В этой реакции молекула спирта выступает в роли нуклеофильного агента, атакующего бедный электронами углеродный атом карбонильной группы.
Реакции этерификации обратимы и, следовательно, ограничены состоянием равновесия. Превращение эквимолекулярных количеств кислоты и спирта в теоретически вычисленное количество сложного эфира по причине обратимости реакции невозможно. В результате реакции образуется некоторое максимальное количество эфира (которое всегда ниже теоретического) и остаются непрореагировавшие спирт и кислота. Например, при нагревании с обратным холодильником эквимолекулярных количеств уксусной кислоты и этилового спирта в реакцию вступает лишь 2/3 г-мол каждого компонента, поэтому максимальный выход эфира в этих условиях составляет лишь 2/3 теоретического, т. е. 66,7%.
/>
По мере того как кислота и спирт реагируют друг с другом и происходит накопление продуктов их взаимодействия (эфира и воды), скорость обратной реакции, вначале незначительная, возрастает. При этом скорость прямой реакции постепенно уменьшается. Наконец, наступает динамическое равновесие, когда в единицу времени в сложный эфир превращается столько же молекул кислоты и спирта, сколько молекул сложного эфира распадается на кислоту и спирт. Одинаковой скоростью этих противоположно протекающих процессов обусловлен постоянный состав системы. Поскольку скорость бимолекулярной реакции пропорциональна произведению концентраций реагирующих веществ, мы можем для скоростей прямой и обратной реакций написать уравнения:
/>
где v1— скорость реакции этерификации; v2— скорость реакции гидролиза; К1и К2— константы скорости обеих реакций; Ск, Сс, Сэи Св— концентрации реагирующих и получающихся веществ (кислоты, спирта, эфира, воды).
В состоянии равновесия скорости реакций, протекающих в противоположных направлениях, равны, т. е. V1= V2. Тогда К1СкСс= КгСэСвили:
/>
Частное К2/К1является константой равновесия и обозначается буквой К.
Из полученного уравнения следует, что в состоянии равновесия отношение произведений концентраций реагирующих веществ обратно отношению констант скоростей реакций. В случае реакции образования уксусноэтилового эфира в состоянии равновесия, как упомянуто выше, в реакционной смеси содержится по 1/3 моля кислоты и спирта и по 2/3 моля эфира и воды. Поэтому
/>
Однако можно изменить состояние равновесия и повысить выход сложного эфира, увеличивая концентрацию спирта (или кислоты). Например, если взять уксусную кислоту и спирт в молярном отношении, равном 1:2, выход эфира (из расчета на кислоту) повышается до 85%. Действительно, пусть концентрация эфира в состоянии равновесия (в молях) будет равна х, т. е. Сэ= х. Тогда и Св= х. Концентрация кислоты Ск= 1—х, концентрация спирта Сс= 2 — х. Следовательно,
/>
После решения этого уравнения находим, что х = 0,85 моля, то есть выход эфира равен 85% теоретического.
Часто применяется и другой способ смещения равновесия в сторону большего выхода сложного эфира — удаление сложного эфира или воды из сферы реакции. Легко можно видеть, что уменьшение концентраций эфира или воды влечет уменьшение концентраций спирта и кислоты, поскольку величина константы равновесия К при данной температуре неизменна. Так, в случае получения низкокипящих сложных эфиров (например, уксусно-этилового с температурой кипения 77°С) в ходе реакции отгоняют эфир из реакционной колбы. При получении высококипящих сложных эфиров (например, уксуснобутилового с температурой кипения 125°С или уксусноизоамилового с температурой кипения 142°С) удобнее отгонять воду в процессе реакции. Вода в этом случае отгоняется в виде азеотропа с парами соответствующего спирта. При конденсации паров в холодильнике происходит расслоение этих ограниченно смешивающихся жидкостей и вода, как более тяжелая, собирается на дне поставленной на пути конденсата «ловушки» (см. рис. 27). Азеотропную отгонку воды можно использовать и в случае этерификации кислот этиловым или пропиловым спиртом, которые в жидкой фазе смешиваются с водой во всех отношениях. В этом случае для отделения воды от сконденсировавшегося в холодильнике спирта в реакционную смесь приходится добавлять третий компонент, образующий с водой и спиртом нераздельно кипящую смесь, но в жидкой фазе с водой не смешивающийся. Его роль состоит в том, что он экстрагирует из конденсата спирт и возвращает его в реакционный сосуд. В качестве такого компонента могут использоваться бензол, хлороформ, четыреххлористый углерод и некоторые другие жидкости, но из перечисленных только бензол можно использовать в «ловушках». Хлороформ и четыреххлористый углерод обладают большей плотностью, чем вода, и для отделения воды от реакционной смеси в случае использования этих жидкостей требуется «ловушка» другой конструкции.
продолжение --PAGE_BREAK--При комнатной температуре реакция протекает очень медленно. При смешении эквимолярных количеств спирта и кислоты для достижения равновесных концентраций требуется до 16 лет. Повышение температуры ускоряет реакцию (так, в случае взаимодействия этилового спирта с уксусной кислотой при 110°Сравновесие достигается через 10 дней, а при (155°С— через несколько часов).
Особенно сильное ускорение реакции этерификации достигается применением катализаторов — водородных ионов, получающихся при диссоциации сильных минеральных кислот. В качестве катализаторов чаще всего используются концентрированная серная кислота или сухой хлористый водород, ток которого пропускается через реакционную смесь. Найдено, что скорость реакции возрастает с увеличением количества катализатора; однако известно также, что добавка 0,01% серной кислоты достаточна для образования этилацетата из спирта и уксусной кислоты. Следует иметь в виду, что катализаторы повышают скорость реакции этерификации, но не могут вызывать сдвига равновесия.
Карбоновые кислоты, как видно из вышесказанного, реагируют со спиртами относительно медленно. Это объясняется слабой активностью карбонильной группы в кислотах по отношению к нуклеофильным агентам по сравнению с активностью той же группы в ангидридах и хлорангидридах кислот, поскольку +М-эффект гидроксильной группы приводит к уменьшению положительного заряда карбонильного углерода
/>
Скорость этерификации карбоновой кислоты тем выше, чем больше положительный заряд карбонильного углерода. Величина δ+ на углероде карбоксильной группы зависит от характера радикала кислоты. Электронодонорные группы, связанные с карбоксилом, понижают дробный положительный заряд (по сравнению с зарядом в муравьиной кислоте) и тем препятствуют взаимодействию кислоты с нуклеофилом; электроноакцепторные заместители, напротив, делают кислоту более реакционноспособной. Поэтому кислоты типа трихлоруксусной, щавелевой, муравьиной быстро реагируют со спиртами даже без добавок минеральной кислоты-катализатора, а ароматические кислоты, особенно те, которые в ароматическом ядре содержат электронодонорные заместители, взаимодействуют со спиртом значительно труднее и требуют больших количеств катализатора.
Сильное влияние на скорость реакции этерификации оказывают также пространственные факторы. С увеличением объема связанных с карбоксилом углеводородных радикалов и с повышением объема этерифицируемых спиртов скорость этерификации уменьшается. Среди спиртов одного молекулярного веса быстрее всего взаимодействуют с кислотами первичные, медленнее — третичные спирты.
Реакцию этерификации можно проводить и в паровой фазе над твердыми катализаторами. Пары спирта и кислоты при 280—300° С пропускают через трубку с катализатором (ThO2 или TiO2). Выходы сложных эфиров в этом случае такие же, как и при реакциях в гомогенной фазе.
Аминокислоты образуют сложные эфиры при взаимодействии со спиртами в присутствии сухого хлористого водорода. Роль хлористого водорода здесь не ограничивается катализом реакции или сдвигом равновесия за счет связывания воды. В присутствии хлористого водорода аминокислота, находившаяся ранее в форме внутренней соли, превращается в хлористоводородную соль аминокислоты, причем карбоксильная группа из неактивной формы аниона переходит в реакционноспособную форму —СООН:
/>
В результате этерификации в этих условиях эфиры также получаются в виде солей. Например, из аминоуксусной кислоты (гликоколя) и абсолютного этилового спирта образуется хлористоводородная соль эфира гликоколя
/>
Свободный эфир из соли можно получить, удаляя хлористый водород окисью серебра:
/>
Механизм этерификации
Роль катализатора заключается в протонировании карбонильного кислорода: при этом карбонильный атом углерода становится более положительным и более «уязвимым» по отношению к атаке нуклеофильного агента, которым является молекула спирта. Образующийся вначале катион (VIII) присоединяет молекулу спирта за счет неподеленных электронов кислородного атома, давая катион (IX):
/>
Далее катион (IX) отщепляет молекулу воды, превращаясь в катион сложного эфира (X):
/>
Катион (X) в результате отщепления протона образует молекулу сложного эфира:
/>
Использование метода «меченых атомов» дало возможность решить вопрос о месте разрыва связей при реакции этерификации. Оказалось, что обычно молекула воды образуется из гидроксила кислоты и водорода спирта. Следовательно, в молекуле кислоты разрывается связь между ацилом и гидроксилом, а в молекуле спирта — связь водорода с кислородом. Такой именно вывод следует из результатов работы по этерификации бензойной кислоты метанолом, содержащим тяжелый изотоп кислорода О18. Полученный сложный эфир содержал в своем составе указанный изотоп кислорода:
Присутствие О18установлено сжиганием образца эфира и анализом образующихся продуктов сгорания (CO2и Н2О) на присутствие тяжелого изотопа кислорода.
Гидролиз сложных эфиров представляет собой реакцию, обратную реакции их образования. Гидролиз может быть осуществлен как в кислой, так и в щелочной среде. Для кислого гидролиза сложных эфиров справедливо все, что было сказано выше применительно к реакции этерификации, об обратимости и механизме процесса, о методах смещения равновесия. Щелочной гидролиз сложных эфиров проходит через следующие стадии:
/>
Он является процессом необратимым, поскольку богатый электронами анион кислоты не способен взаимодействовать с нуклеофильной молекулой спирта.
Практически щелочной гидролиз сложных эфиров проводят в присутствии едких щелочей КОН, NaOH, а также гидроокисей щелочноземельных металлов Ва(ОН)2, Са(ОН)2Образующиеся при гидролизе кислоты связываются в виде солей соответствующих металлов, поэтому гидроокиси приходится брать по крайней мере в эквивалентном отношении со сложным эфиром. Обычно используют избыток основания. Выделение кислот из их солей осуществляется с помощью сильных минеральных кислот.
В качестве растворителя основания для реакции гидролиза чаще всего применяют воду, которая, однако, не растворяет сложный эфир. Реакция идет на поверхности раздела двух фаз и требует поэтому хорошего перемешивания. Иногда реакцию бывает целесообразно проводить в гомогенной среде, используя в качестве растворителя водный спирт. При этом, однако, нужно иметь в виду, что для выделения кислоты перед подкислением раствора спирт необходимо удалить (отогнать).
Список литературы
Общая органическая химия. Карбоновые кислоты и их производные. Том 4. М., Химия, 1983, 729с.
Шабаров Ю.С. Органическая химия: В 2-х кн. — М.: Химия, 1994.- 848 с.
Петров А.А., Бальян Х.В., Трощенко А.Т. Органическая химия. – М.: Высш. шк., 1973. — 623 с.
Препаративная органическая химия. Изд. 2-е, М., Госхимиздат, 1964.
Богословский Б.Н., Казакова З.С. Скелетные катализаторы, их свойства и применение в органической химии. М., Госхимиздат, 1957.
Голодников Г.В. Практические работы по органическому синтезу. Л., Изд-во ЛГУ, 1966, 697с.
Дорофеенко Г.Н., Жданов Ю.А., Дуленко В.И. и др. Хлорная кислота и ее соединения ворганическом синтезе. Ростов, изд-во Ростовского ун-та, 1965.
Терней А. Современная органическая химия: В 2 т. — М.: Мир, 1981. — Т.1 — 670 с; Т.2 — 615 с.
Лабораторные работы по органической химии. Изд. 3-е. М., Высшая школа, 1974.
Храмкина М.Н. Практикум по органическому синтезу. Изд. 4-ое, Л., Химия. 1977.
В. Ф. Травень. Органическая химия. Том 1. – М.: Академкнига, 2004, — 708 с.
Голодников Г.В., Низовкина Т.В., Рыскальчук А.Т. Практикум по органическому синтезу. Л., Изд-во ЛГУ, 1967.
Крешков А.П., Курбатов И.Н. Лабораторные работы по синтезу и анализу органических соеднений. М., изд-во Артиллерийского ордена Ленина академии Красной армии им. Дзержинского, 1940.
www.ronl.ru