Способ получения диэтилового эфира итаконовой кислоты. Технология получения диэтилового эфира
Способ выделения диэтилового эфира
Изобретение относится к способу выделения диэтилового эфира из эфироальдегидной фракции - отхода производства синтетического этилового спирта. Способ включает взаимодействие эфироальдегидной фракции с перекисью водорода в присутствии каталитического количества сильной кислоты и последующее выделение целевого продукта ректификацией. Как правило, кислоту используют в количестве 0,001-0,1 мас. %. Способ позволяет сократить время проведения процесса. 1 з.п. ф-лы, 1 табл.
Изобретение относится к химической промышленности, а именно к выделению диэтилового эфира из эфироальдегидной фракции отхода производства синтетического этилового спирта. Эфироальдегидная фракция имеет преимущественно следующий состав, мас. %: Диэтиловый эфир - 55-80 Ацетальдегид - 5-25 Ацетон - До 1 Метилэтилкетон - До 0,2 Этиловый спирт - Остальное (Технические условия АО "Уфаоргсинтез" ТУ 2434-034-05766563-96 "Фракция Эфироальдегидная") Диэтиловый эфир и ацетальдегид, являющиеся основными компонентами эфироальдегидной фракции, имеют близкие температуры кипения, вследствие этого их разделение представляет определенные трудности и является одной из причин того, что эфироальдегидную фракцию сжигают. Между тем эта фракция содержит более 50% диэтилового эфира, который находит широкое применение в имической промышленности и медицине.
РИСУНКИ
Рисунок 1www.findpatent.ru
Способ получения диэтилового эфира фталевой кислоты
Предлагаемое изобретение относится к получению эфиров карбоновых кислот, а именно к получению диэтилового эфира фталевой кислоты - диэтилфталата. Способ получения диэтилфталата заключается во взаимодействии фталевого ангидрида и этилового спирта в пленочном режиме в присутствии твердого катализатора -окиси алюминия, пропитанной серным ангидридом не менее 15% при температуре 120-130oС. Технический результат - упрощение процесса. 2 з.п. ф-лы, 1 табл., 1 ил.
Изобретение относится к области органической химии, в частности к получению эфиров карбоновых кислот, содержащих этерифицированную карбоксильную группу, связанную с атомом углерода шестичленного ароматического ядра, а именно к получению диэтилового эфира фталевой кислоты - диэтилфталата. Диэтилфталат широко используется в качестве пластификатора пластмасс, растворителя и в различных областях парфюмерии, косметики и медицины.
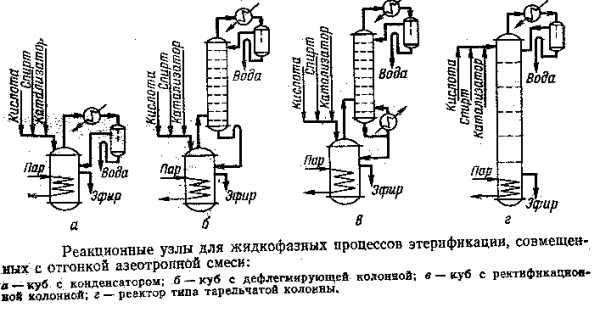
Известны способы получения диэтилфталата взаимодействием этилового спирта с фталевым ангидридом, или фталевой кислотой или ее хлорангидридом. Известен способ получения диэтилфталата взаимодействием фталевого ангидрида с этиловым спиртом в присутствии катализатора серной кислоты [1]. Процесс проводят пропусканием паров спирта через реакционную смесь фталевого ангидрида и серной кислоты при температуре 120-130oC. При этом диэтилфталат остается в расплаве фталевого ангидрида, а пары воды и пары непрореагировавшего этилового спирта конденсируются и собираются в виде водного раствора. Полученный диэтилфталат содержит от 3 до 5% серной кислоты. Далее диэтилфталат-сырец отмывают от серной кислоты и сушат. Недостатками этого способа являются: высокий расход этилового спирта, возврат которого в процесс осуществляют после обезвоживания; отмывка диэтилфталата-сырца от серной кислоты; в качестве отходов большое количество сточных вод. Задачей настоящего изобретения является выявление более оптимальных условий проведения этерификации фталевого ангидрида парами этилового спирта, создание более простого технологического процесса, используемого в промышленном производстве с большим экономическим эффектом. Для решения поставленной задачи предлагается проводить этерификацию фталевого ангидрида парами этилового спирта в пленочном режиме в присутствии твердого катализатора - гранулированной -окиси алюминия, пропитанного серным ангидридом не менее 15%. Процесс проводят при температуре 120-130oC. Использование твердого катализатора - гранулированной - окиси алюминия, пропитанного серным ангидридом, позволяет использовать твердый катализатор одновременно и как насадку, которая обеспечивает взаимодействие фталевого ангидрида с этиловым спиртом в пленочном режиме. Это позволяет избежать недостатков, которые присутствуют при использовании жидкого катализатора - концентрированной серной кислоты в способе прототипе. Предлагаемое техническое решение позволяет упростить технологический процесс получения диэтилфталата и сделать его более экономичным в промышленном производстве при получении высоких конечных результатов товарной продукции. Способ отработан в опытном производстве при непрерывном режиме. Пример Для проведения способа получения диэтилфталата взаимодействием фталевого ангидрида с этиловым спиртом в присутствии твердого катализатора - гранулированной -Al2O3, пропитанного серным ангидридом, предварительно приготовляют катализатор этерификации. В цилиндрический аппарат помещают гранулированный - Al2O3, предварительно высушенный и освобожденный от пыли рассеиванием. Через аппарат, содержащий - Al2O3, пропускают контролируемый поток сухого воздуха, содержащего 6-7% мас. серного ангидрида. Количество пропускаемого воздуха определяют из расчета полного поглощения серного ангидрида оксидом алюминия в массовом соотношении Al2O3:SO3 = 15:1. Подготовленный таким образом катализатор выгружают в сухой атмосфере, взвешивают и перегружают в реактор колонного типа для этерификации, который представлен на чертеже. Цилиндрический реактор изготовлен из нержавеющей стали, внутренний диаметр реактора 6,0 см. В реактор загружают катализатор высотой 60 см, объем расположенного в реакторе катализатора 1,7 дм3. В реактор, в верхнюю его часть через штуцер A дозируют расплавленный фталевый ангидрид (To = 130-140oC). Под слой расплавленного фталевого ангидрида дозируют этиловый спирт-ректификат. Необходимую температуру в реакторе (13010oC) поддерживают электронагреватели (обмотки), расположенные на внешней стенке реактора. Образующиеся пары этилового спирта взаимодействуют с расплавленным фталевым ангидридом при течении его по катализатору-насадке в виде пленки. Процесс протекает по реакции: C6h5(CO)2O + 2C2H5OH ---> C6h5(COOC2H5)2 + h3O Образующийся диэтилфталат стекает в нижнюю часть реактора и после охлаждения поступает в сборник товарного продукта, а парообразные продукты реакции - вода и непрореагировавший этиловый спирт - выводят из реактора через штуцер D, расположенный в нижней части реактора. Пленочный режим в виде падающей пленки по поверхности катализатора-насадки обеспечивает наилучший контакт реагирующих компонентов, находящихся в двух фазах, и свободный вывод жидких и газообразных продуктов реакции. Для расплавления фталевого ангидрида, его дозирования, дозирования этилового ангидрида, конденсирования парообразных продуктов реакции используют стандартное оборудование и приборы. Для сравнения в этом же реакторе были проведены опыты по получению диэтилфталата взаимодействием фталевого ангидрида с этиловым спиртом в присутствии катализатора - гранулированной - окиси алюминия, пропитанного серным ангидридом в барботажном режиме. В реакторе (см. чертеж) для создания барботажного режима изменяли поток реагентов на восходящий. Расплавленный фталевый ангидрид и этиловый спирт дозировали через штуцеры C и D, расположенные в нижней части реактора. Через штуцер A и штуцер B, расположенные в верхней части реактора, осуществляли вывод жидкой реакционной смеси, пары воды и этилового спирта. При проведении процесса этерификации скорость и время дозирования фталевого ангидрида и этилового спирта определяли при помощи насоса-дозатора и часов. Отбор проб реакционных смесей проводили после прохождения через реактор 1,5 л суммарной реакционной смеси фталевого ангидрида и этилового спирта для обеспечения выхода процесса на стационарный режим. Содержание компонентов реакционной смеси определяли методом газожидкостной хроматографии (ГЖХ). Технологические параметры проведения опытов представлены в таблице. Из таблицы видно, что проведение этерификации фталевого ангидрида этиловым спиртом в присутствии твердого катализатора - гранулированной Al2O3, пропитанной серным ангидридом в пленочном режиме, имеет значительные преимущества перед барботажным режимом по конечным результатам процесса: выходу диэтилфталата, его содержанию в готовом продукте, а также содержанию этилового спирта в конденсате. Предлагаемый способ получения диэтилфталата более прост, технологичен и экономичен. Источники: 1. Масложировая промышленность, 1985, N 5 "Получение метиловых и этиловых эфиров высококипящих карбоновых кислот".www.findpatent.ru
способ получения диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты - патент РФ 2446149
Изобретение относится к улучшенному способу получения диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты. Способ заключается в циклоконденсации диэтилового эфира ацетондикарбоновой кислоты с изонитрозоацетилацетоном с последующим восстановлением полученного диэтилового эфира 2-гидрокси-4,6-диметил-5-нитрозоизофталевой кислоты водородом на палладиевом катализаторе в среде этилацетата до получения конечного продукта. Способ позволяет сократить число стадий процесса, повысить выход целевого продукта и сделать процесс более безопасным. 1 пр.
Изобретение относится к органической химии, в частности к способу синтеза ароматического соединения: диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты, который используется как полупродукт для получения фармацевтических препаратов.
Известно, что производные диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты - это соединения, представляющие большой практический интерес, так как они являются основой современных противоаритмических препаратов / 5-Aminoacetamido-4,6-dimethyl-2-hydroxyisophtalic acid diethyl ester: synthesis and study of antiarrhythmic properties // Von F. Eiden, H.P.Leister, D.Mayer / Arzneimittel-Forschung, 1983, 33(1), 101-105.
Диэтиловый эфир 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты применяется для получения на его основе противоаритмических препаратов, которые в настоящее время выпускаются в Китае.
Известен единственный способ получения диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты. Его получают по следующей схеме: из диэтилового эфира ацетондикарбоновой кислоты получают его динатриевую соль, затем ацилируют хлористым ацетилом до образования диацетилпроизводного, а из него получают 3,5-диэтоксикарбонил-2,6-диметил-4-пирон / Ricerche dirette alla sintesi del pirone. Nuovo modo di formazione dell'etere dimetilpirondicarbonico // di A. Peratoner e B. Strazzeri / Gazzetta chimica italiana, 1891, 21, 292-300:
Затем полученный 3,5-диэтоксикарбонил-2,6-диметил-4-пирон обрабатывают нитрометаном в среде третбутилата калия в атмосфере азота. Образуется диэтиловый эфир 2-гидрокси-4,6-диметил-5-нитроизофталевой кислоты, который гидрируют водородом на катализаторе, получая целевой продукт: диэтиловый эфир 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты / 5-Aminoacetamido-4,6-dimethyl-2-hydroxyisophtalic acid diethyl ester: synthesis and study of antiarrhythmic properties // Von F. Eiden, H.P.Leister, D.Mayer / Arzneimittel-Forschung, 1983, 33(1), 101-105:
Недостатками известного метода является то, что пятистадийный синтез протекает с суммарным низким выходом - 14,3%. При этом четвертая стадия проводится в третбутилате калия с нитрометаном, это трудоемко и взрывоопасно, так как третбутилат калия получают из металлического калия и третичного бутилового спирта, обычно в атмосфере азота. В результате образуется производное нитрофенола - диэтиловый эфир 2-гидрокси-4,6-диметил-5-нитроизофталевой кислоты, затем конечный продукт - диэтиловый эфир 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты, на получение которого расходуется значительное количество водорода и времени (3 дня).
Изобретение решает задачу разработки нового способа получения диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты, более технологичного с меньшим количеством стадий, безопасного и большим суммарным выходом.
Технический результат заключается в создании нового способа получения диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты, более простого и технологичного, с меньшим количеством стадий, безопасного, с высоким выходом. Суммарный выход конечного продукта 62,4%, тогда как в прототипе 14,3%.
Изобретательский уровень данного способа определяется тем, что исходя из того же исходного вещества предложена новая последовательность реакций, приводящая к образованию диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты. Впервые диэтиловый эфир 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты получают через образование перзамещенного пара-нитрозофенола путем циклизации диэтилового эфира ацетондикарбоновой кислоты с изонитрозоацетилацетоном и последующим восстановлением образующегося продукта, что позволяет сократить число стадий, повысить выход и сделать процесс более безопасным.
Указанный технический результат достигается тем, что способ получения диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты осуществляется циклоконденсацией диэтилового эфира ацетондикарбоновой кислоты с изонитрозоацетилацетоном и последующим восстановлением полученного диэтилового эфира 2-гидрокси-4,6-диметил-5-нитрозоизофталевой кислоты водородом на палладиевом катализаторе в среде этилацетата с получением конечного продукта.
Диэтиловый эфир 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты получается по следующей схеме:
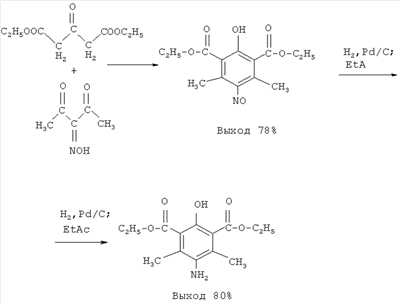
Преимущество данного способа заключается в том, что процесс получения диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты осуществляется из того же исходного соединения, что и в прототипе, но всего в две стадии, причем обе проводятся при комнатной температуре, то есть в значительно более мягких условиях, чем в известном способе. Кроме того, стадия восстановления требует вдвое меньшего количества водорода и значительно меньшего времени гидрирования (несколько часов), чем в известном способе, так как восстановлению подвергается нитрозогруппа, в которой только один атом кислорода, а не два, как в нитрогруппе.
Пример. Сначала получают диэтиловый эфир 2-гидрокси-4,6-диметил-5-нитрозоизофталевой кислоты общей формулы
циклоконденсацией изонитрозоацетилацетона с диэтиловым эфиром ацетондикарбоновой кислоты.
Растворяют 5 ммоль гидроксида калия в 5 мл абсолютного спирта и добавляют 5 ммоль изонитрозоацетилацетона и 10 ммоль диэтилового эфира ацетондикарбоновой кислоты. Смесь выдерживают 20 мин при 18-20°С. Раствор менял цвет с оранжевого до зеленого. Калиевую соль выделяют добавлением абсолютного диэтилового эфира до помутнения. Фильтруют ярко-зеленые кристаллы, сушат в вакууме над Na 2SO4. Для выделения димерного нитрозофенола полученную соль растворяли в 4 мл воды, подкисляли разбавленной соляной кислотой (1:3). При этом выпадало маслообразное вещество, которое быстро затвердевало в бесцветные кристаллы. Получают белые кристаллы, т.пл. 103-105°С. Спектр ЯМР 1 H калиевой соли (D2O), , м.д.: 1.30 т (6Н, 2СН3), 2.35 с (6Н, 2СН 3), 4.34 к (4Н, 2СН2). Масс-спектр, m/z (I отн., %): 293 (25), 247 (50), 221 (10), 202 (50), 175 (100), 147 (10), 119 (10), 91 (15), 67 (10), 51 (10).
Затем полученный диэтиловый эфир 2-гидрокси-4,6-диметил-5-нитрозоизофталевой кислоты гидрируют в статической системе при атмосферном давлении в термостатируемой стеклянной ячейке объемом 20 мл при перемешивании и 298±0.1 К. В ячейку помещают суспензию 0.1 г катализатора - 0,5% Pd/C в виде порошка в 5 мл этилацетата. Систему откачивают и заполняют водородом последовательно пять раз, затем раствор 0,5 ммолей нитрозофенола в 5 мл этилацетата вводят в реактор в токе водорода и включают перемешивание. Изменение режима перемешивания не влияет на скорость гидрирования, при этом скорость поглощения водорода не превышала 3 мл/мин. Время гидрирования составило 100 минут. После полного поглощения водорода отфильтровывают реакционную массу от катализатора и насыщают сухим хлороводородом, в результате чего выпадает гидрохлорид диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты в виде белых кристаллов. Осадок отфильтровывают и промывают толуолом. Получают белые кристаллы хлоргидрата, т.пл. 168-170°С, из которого при подщелачивании выделяют чистый диэтиловый эфир 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты с т.пл. 72-74°С. Спектр ЯМР 1Н (ацетон-d6), , м.д.: 1.43 т (6Н, 2СН3), 2.39 с (6Н, 2СН 3), 3.01 уш. с (2Н, Nh3), 4.49 к (4Н, 2СН 2), 11.55 с (1Н, ОН). Масс-спектр, m/z (Iотн ., %): 281 (50) [М]+, 235 (100), 207 (15), 189 (85), 163 (30), 133 (20), 106 (30), 77 (15), 53 (10). В УФ-спектре в этаноле 96% присутствует максимум в области 352,5 нм ( =4420). В ИК-спектре присутствует широкая полоса поглощения аминогруппы в области 3300-3500 см-1 и две сильные полосы поглощения карбонильных групп в области 1673 см-1 и 1727 см-1.
Способ получения диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты отличается простой двухстадийной схемой синтеза с выходом, значительно превышающим выход того же продукта, получаемого по известному способу.
ФОРМУЛА ИЗОБРЕТЕНИЯ
Способ получения диэтилового эфира 5-амино-2-гидрокси-4,6-диметилизофталевой кислоты из диэтилового эфира ацетондикарбоновой кислоты, отличающийся тем, что диэтиловый эфир ацетондикарбоновой кислоты подвергают циклоконденсации с изонитрозоацетилацетоном с последующим восстановлением полученного диэтилового эфира 2-гидрокси-4,6-диметил-5-нитрозоизофталевой кислоты водородом на палладиевом катализаторе в среде этилацетата до получения конечного продукта.
www.freepatent.ru
Способ получения диэтилового эфира фталевой кислоты
,
ОПИСАНИЕ ИЗОБРЕТЕНИЯ К ПАТЕНТУ
Изобретение относится к области органической химии, в частности к получению эфиров карбоновых кислот, содержащих этерифицированную карбоксильную группу, связанную с атомом углерода шестичленного ароматического ядра, а именно к получению диэтилового эфира фталевой кислоты - диэтилфталата. Диэтилфталат широко используется в качестве пластификатора пластмасс, растворителя и в различных областях парфюмерии, косметики и медицины. Известны способы получения диэтилфталата взаимодействием этилового спирта с фталевым ангидридом, или фталевой кислотой или ее хлорангидридом. Известен способ получения диэтилфталата взаимодействием фталевого ангидрида с этиловым спиртом в присутствии катализатора серной кислоты [1]. Процесс проводят пропусканием паров спирта через реакционную смесь фталевого ангидрида и серной кислоты при температуре 120-130oC. При этом диэтилфталат остается в расплаве фталевого ангидрида, а пары воды и пары непрореагировавшего этилового спирта конденсируются и собираются в виде водного раствора. Полученный диэтилфталат содержит от 3 до 5% серной кислоты. Далее диэтилфталат-сырец отмывают от серной кислоты и сушат. Недостатками этого способа являются: высокий расход этилового спирта, возврат которого в процесс осуществляют после обезвоживания; отмывка диэтилфталата-сырца от серной кислоты; в качестве отходов большое количество сточных вод. Задачей настоящего изобретения является выявление более оптимальных условий проведения этерификации фталевого ангидрида парами этилового спирта, создание более простого технологического процесса, используемого в промышленном производстве с большим экономическим эффектом. Для решения поставленной задачи предлагается проводить этерификацию фталевого ангидрида парами этилового спирта в пленочном режиме в присутствии твердого катализатора - гранулированной γ-окиси алюминия, пропитанного серным ангидридом не менее 15%. Процесс проводят при температуре 120-130oC. Использование твердого катализатора - гранулированной γ- окиси алюминия, пропитанного серным ангидридом, позволяет использовать твердый катализатор одновременно и как насадку, которая обеспечивает взаимодействие фталевого ангидрида с этиловым спиртом в пленочном режиме. Это позволяет избежать недостатков, которые присутствуют при использовании жидкого катализатора - концентрированной серной кислоты в способе прототипе. Предлагаемое техническое решение позволяет упростить технологический процесс получения диэтилфталата и сделать его более экономичным в промышленном производстве при получении высоких конечных результатов товарной продукции. Способ отработан в опытном производстве при непрерывном режиме. Пример Для проведения способа получения диэтилфталата взаимодействием фталевого ангидрида с этиловым спиртом в присутствии твердого катализатора - гранулированной γ-Al2O3, пропитанного серным ангидридом, предварительно приготовляют катализатор этерификации. В цилиндрический аппарат помещают гранулированный γ- Al2O3, предварительно высушенный и освобожденный от пыли рассеиванием. Через аппарат, содержащий γ- Al2O3, пропускают контролируемый поток сухого воздуха, содержащего 6-7% мас. серного ангидрида. Количество пропускаемого воздуха определяют из расчета полного поглощения серного ангидрида оксидом алюминия в массовом соотношении Al2O3:SO3 = 15:1. Подготовленный таким образом катализатор выгружают в сухой атмосфере, взвешивают и перегружают в реактор колонного типа для этерификации, который представлен на чертеже. Цилиндрический реактор изготовлен из нержавеющей стали, внутренний диаметр реактора 6,0 см. В реактор загружают катализатор высотой 60 см, объем расположенного в реакторе катализатора 1,7 дм3. В реактор, в верхнюю его часть через штуцер A дозируют расплавленный фталевый ангидрид (To = 130-140oC). Под слой расплавленного фталевого ангидрида дозируют этиловый спирт-ректификат. Необходимую температуру в реакторе (130±10oC) поддерживают электронагреватели (обмотки), расположенные на внешней стенке реактора. Образующиеся пары этилового спирта взаимодействуют с расплавленным фталевым ангидридом при течении его по катализатору-насадке в виде пленки. Процесс протекает по реакции: C6h5(CO)2O + 2C2H5OH ---> C6h5(COOC2H5)2 + h3O Образующийся диэтилфталат стекает в нижнюю часть реактора и после охлаждения поступает в сборник товарного продукта, а парообразные продукты реакции - вода и непрореагировавший этиловый спирт - выводят из реактора через штуцер D, расположенный в нижней части реактора. Пленочный режим в виде падающей пленки по поверхности катализатора-насадки обеспечивает наилучший контакт реагирующих компонентов, находящихся в двух фазах, и свободный вывод жидких и газообразных продуктов реакции. Для расплавления фталевого ангидрида, его дозирования, дозирования этилового ангидрида, конденсирования парообразных продуктов реакции используют стандартное оборудование и приборы. Для сравнения в этом же реакторе были проведены опыты по получению диэтилфталата взаимодействием фталевого ангидрида с этиловым спиртом в присутствии катализатора - гранулированной γ- окиси алюминия, пропитанного серным ангидридом в барботажном режиме. В реакторе (см. чертеж) для создания барботажного режима изменяли поток реагентов на восходящий. Расплавленный фталевый ангидрид и этиловый спирт дозировали через штуцеры C и D, расположенные в нижней части реактора. Через штуцер A и штуцер B, расположенные в верхней части реактора, осуществляли вывод жидкой реакционной смеси, пары воды и этилового спирта. При проведении процесса этерификации скорость и время дозирования фталевого ангидрида и этилового спирта определяли при помощи насоса-дозатора и часов. Отбор проб реакционных смесей проводили после прохождения через реактор 1,5 л суммарной реакционной смеси фталевого ангидрида и этилового спирта для обеспечения выхода процесса на стационарный режим. Содержание компонентов реакционной смеси определяли методом газожидкостной хроматографии (ГЖХ). Технологические параметры проведения опытов представлены в таблице. Из таблицы видно, что проведение этерификации фталевого ангидрида этиловым спиртом в присутствии твердого катализатора - гранулированной γ Al2O3, пропитанной серным ангидридом в пленочном режиме, имеет значительные преимущества перед барботажным режимом по конечным результатам процесса: выходу диэтилфталата, его содержанию в готовом продукте, а также содержанию этилового спирта в конденсате. Предлагаемый способ получения диэтилфталата более прост, технологичен и экономичен. Источники: 1. Масложировая промышленность, 1985, N 5 "Получение метиловых и этиловых эфиров высококипящих карбоновых кислот".ФОРМУЛА ИЗОБРЕТЕНИЯ
1. Способ получения диэтилового эфира фталевой кислоты этерификацией фталевого ангидрида этиловым спиртом в присутствии катализатора при повышенной температуре, отличающийся тем, что взаимодействие фталевого ангидрида с этиловым спиртом проводят в пленочном режиме, в качестве катализатора используют гранулированную γ-окись алюминия, пропитанную серным ангидридом. 2. Способ по п. 1, отличающийся тем, что взаимодействие фталевого ангидрида с этиловым спиртом проводят при 120 - 130oC. 3. Способ по п.1, отличающийся тем, что гранулированная γ-окись алюминия содержит не менее 15% серного ангидрида.bankpatentov.ru
Способ выделения диэтилового эфира
Изобретение относится к химической промышленности, к утилизации отходов производства синтетического этилового спирта, а именно к выделению диэтилового эфира из эфироальдегидной фракции. При этом эфироальдегидную фракцию обрабатывают перекисью водорода, взятой в эквимолярном количестве или в небольшом избытке к содержащемуся в эфироальдегидной фракции ацетальдегиду, а затем ректификацией выделяют диэтиловый эфир. Технический результат - получение диэтилового эфира с содержанием основного вещества не менее 99,5%, пригодного для использования в химической промышленности и медицине. 2 з.п. ф-лы.
Изобретение относится к химической промышленности, к утилизации отходов производства синтетического этилового спирта, а именно к выделению диэтилового эфира из эфироальдегидной фракции.
Эфироальдегидная фракция является отходом производства синтетического этилового спирта, образуется в процессе его обезвоживания, преимущественно имеет следующий состав, мас.%: Диэтиловый эфир - 55-80 Ацетальдегид - 5-25 Примеси: Ацетон - До 1 Метилэтилкетон - до 0,2 Этиловый спирт - Остальное Технические условия АО "Уфаоргсинтез" ТУ 2434-034-05766563-96 "Фракция эфироальдегидная" Диэтиловый эфир и ацетальдегид, являющиеся основными компонентами эфироальдегидной фракции, имеют близкие температуры кипения (диэтиловый эфир -35,6oC, ацетальдегид-20,8oC), вследствие этого их разделение представляет определенные трудности и является одной из причин того, что эфироальдегидную фракцию выбрасывают в атмосферу или сжигают. Между тем эта фракция содержит более 30% диэтилового эфира, который находит широкое применение химической промышленности и медицине. Известен способ выделения диэтилового эфира из смеси с ацетальдегидом и другими примесями (перекиси, сивушные масла), согласно которому смесь пропускают через колонну с цеолитом натрия, а затем подвергают ректификации с отмывкой водой по принципу противотока (а. с. СССР N 164587, МПК C 07 C 43/06, опубл. 1964) Недостатком указанного способа является его многостадийность и необходимость регенерации абсорбента. Кроме того, указанный способ применим только для очистки технического диэтилового эфира до марки "наркозный" и не годится для выделения диэтилового эфира из смеси, содержащей большое количество других компонентов, какой является эфироальдегидная фракция. Наиболее близким к предлагаемому изобретению является способ очистки технического эфира окислением раствором перманганата калия с последующей обработкой бисульфитом натрия в паровой фазе в колонне непрерывного действия (а. с. СССР N 149104, МПК C 07 C 43/06, опубл. 1962). Недостатком указанного способа является также то, что он не годится для выделения диэтилового эфира из смеси, содержащей большое количество других компонентов. Изобретение решает задачу выделения диэтилового эфира из эфироальдегидной фракции производства этилового спирта. Указанная задача решается тем, что эфироальдегидную фракцию обрабатывают перекисью водорода, (в результате происходит окисление ацетальдегида до уксусной кислоты (температура кипения 118,1oC), а затем ректификацией выделяют диэтиловый эфир. Причем перед введением перекиси водорода смесь эфира и ацетальдегида отделяют от этилового спирта. Перекись водорода берут в эквимолярном количестве или в небольшом избытке к содержанию ацетальдегида. Непрореагировавшую перекись водорода разрушают известным способом, например, добавлением солей железа, например хлорного железа. Пример 1. 1000 кг эфироальдегидной фракции, содержащей 67% диэтилового эфира, 6,5% ацетальдегида, 0,5% ацетона, 0,3% метилэтилкетона, остальное этиловый спирт, подвергли ректификационной перегонке в колонне из 12-ти тарелок с флегмовым числом 1:10. Получили 700 кг дистиллята, содержащего 80% диэтилового эфира, около 19% ацетальдегида и примесей до 1%. В кубе получили этиловый спирт с примесью (4%) диэтилового эфира. В полученный дистиллят добавили при перемешивании 273 кг 38%-ного раствора перекиси водорода, смесь выдержали при комнатной температуре 19 часов, затем добавили при перемешивании 200 г 20%-ного водного раствора хлорного железа, выдержали 20 минут, после чего из полученной смеси в ректификационной колонне отогнали диэтиловый эфир. В результате получили 450 кг диэтилового эфира с содержанием основного вещества 99,5%. В кубе колонны получили уксусную кислоту. Пример 2. Процесс ведут аналогично примеру 1, но для разложения избытка перекиси водорода используют сульфат хрома (III). Получили 448 кг эфира с содержанием основного вещества 99,6%. Пример 3. Процесс ведут аналогично примеру 1, но для разложения избытка перекиси водорода используют сульфат марганца. Получили 449 кг эфира с содержанием основного вещества 99,4%. Изобретение позволяет из отхода производства (эфироальдегидной фракции) получить диэтиловый эфир, пригодный для использования в медицине и химической промышленности. Кроме того, по предложенному способу дополнительно получают уксусную кислоту, также пригодную для использования в химической и пищевой промышленности.Формула изобретения
1. Способ выделения диэтилового эфира из эфироальдегидной фракции производства синтетического спирта путем обработки эфироальдегидной фракции с последующим отделением целевого эфира, отличающийся тем, что обработку осуществляют перекисью водорода, взятой в эквимолярном количестве или в небольшом избытке к содержащемуся в эфироальдегидной фракции ацетальдегиду, а затем ректификацией выделяют диэтиловый эфир. 2. Способ по п.1, отличающийся тем, что перед выделением диэтилового эфира избыток перекиси водорода в реакционной смеси разлагают введением солей металлов. 3. Способ по п.1 или 2, отличающийся тем, что перед введением перекиси водорода эфироальдегидную фракцию отгоняют от этилового спирта.www.findpatent.ru
Фенол, получение с диэтиловым эфиром
Со сколько-нибудь заметной скоростью эта реакция протекает обычно лишь начиная со 150° С. Анизол может быть использован вместо диэтилового эфира при получении реактивов Гриньяра из относительно инертных галоидных производных, поскольку, в анизоле эТот процесс можно проводить при гораздо более высокой температуре. Однако при температуре кипения анизола реактив Гриньяра атакует эфирную связь анизола и образуется фенол [96]. В заметной степени деметилирование фенольных эфиров может происходить уже при температуре кипения бензола, поскольку при взаимодействии дигидротебаина с иодистым изопропилмаг-нием и с бромистым фенилмагнием образуется значительное количество А -енольного метилового эфира дигидроморфинона-6 [99]. [c.376]
Из рассмотрения полученных результатов можно сделать вывод, что универсальная функция межмолекулярного взаимодействия [2, 3] дает хорошее качественное согласие с опытом при описании влияния растворителя на частоту полосы vxн комплекса. Удовлетворительное количественное согласие получается для комплекса метанол + диэтиловый эфир, если принять в качестве радиуса комплекса ван-дер-ваальсовский радиус метанола. Для случая фенола количественное совпадение расчета с экспериментом можно получить, если принять онзагеровский радиус комплекса меньшим ван-дер-ваальсовского радиуса фенола в 1,3 раза. [c.54]Процесс превращения этилового спирта в диэтиловый эфир является примером реакции алкилирования. Превращение это можно осуществить также и в газовой фазе, пропуская пары спирта при температуре 200—300 °С над водоотнимающими катализаторами (оксиды алюминия, титана, тория, ванадия). Из высших спиртов (особенно вторичных и третичных) выход эфиров по описанному способу меньше, чем при получении диэтилового эфира, а фенолы этим путем совсем не образуют простых эфиров. [c.167]
Эпоксидные смолы, полученные из фенолов, растворяются в кетонах, хлорированных углеводородах, диоксане, пиридине, диэтиловом эфире, ароматических углеводородах, ледяной уксусной кислотен не растворяются в воде. Продажные эпоксидные смолы характеризуют по плотности, показателю преломления и вязкости. [c.216]
Очень часто хорошие результаты получают, применяя в качестве растворителя пиридин (иногда хинолин). Этот способ особенно удобен для ацилирования соединений, содержащих несколько гидроксильных групп (многоатомных спиртов, сахаров, фенолов и т. п.). Обычно спирт или фенол растворяют в пиридине, охлаждают раствор и прибавляют к нему хлорангидрид. Через несколько часов стояния при комнатной температуре реакционную смесь, окрашенную в темно-красный цвет, выливают в разбавленную серную кислоту, к которой предварительно добавили немного льда. При этом ацильные производные выделяются в виде масла или в твердом состоянии. Продукт реакции извлекают диэтиловым эфиром или хлороформом и промывают полученную вытяжку разбавленной кислотой для полного удаления пиридина. Если же применяют хлорангидрид плохо растворимой кислоты, то избыток ее удаляют промыванием разбавленным раствором углекислого натрия. [c.164]
Восстановление фенолов.— Другой метод восстановления фенолов в ароматические углеводороды имеет особенно большое значение для химии природных соединений, так как он применим для работы с малыми количествами вещества (50—200 мг) и реакция проводится в таких мягких условиях, что устраняется возможность перегруппировки. Метод был разработан Кеннером (1955) и использован Пелльтье для работы с полумикроколичествами веществ (1958). Раствор фенола и диэтилового эфира фосфористой кислоты в четыреххлористом углероде обрабатывают тризтиламином и оставляют на 24 ч для полного выпадения солянокислого триэтиламина (реакция 1). Затем полученный а.рилдиэтилфосфат отделяют, растворяют в тетрагидрофуране и восстанавливают натрием в жидком аммиаке (реакция 2) [c.187]
Смесь 25,6 г (0,25 М) гидрата окиси рубидия (или 37,5 г (0,25 М) гидрата окиси цезия), не содержащего карбонатов (см. примечания 1,2), 15 мл этилового спирта и 23,5 г (0,255 М) свежеперегнанного фенола помещают в стакан из термостойкого стекла и подогревают при постоянном перемешивании до полного растворения фенола и гидроокиси рубидия (цезия) в спирте. После получения гомогенного раствора его охлаждают до комнатной температуры и выделяют фенолят рубидия (цезия), осаждая его добавлением 150 мл диэтилового эфира. Выделившийся в виде белого кристаллического осадка с перламутровым блеском фенолят рубидия (цезия) отфильтровывают и суп1ат в эксикаторе над едким кали при комнатной температуре. [c.65]
Эфиры фенолов также превращаются о соответствующие альдегиды л аналогичных услоЕШях. Этот метод был с успехом применен для получения альдегидов нз анизола, феиетола, м-хлоранизола, т-хлорфеиетола, дифенилового эфира, вератрола, диэтилового эфира пирокатехина, димети-лового эфира резорцина я а- и р-нафтиловых эфиров. [c.138]
Фенолы предварительно концентрируют, экстрагируя их из подкисленного раствора днэтиловым эфиром или адсорбируя на активированном угле и вымывая затем диэтиловым эфиром. Полученный эфирный раствор фенолов концентрируют в колбочке с оттянутым дном до объема 0,5—1 мл. Отбирают пробу микрошприцом, вводят в испаритель включенного газового хроматографа не более 6 мкл и устанавливают скорость газа-носителя 60 мл/мин, температуру разделения 166— 168° С, ток моста 200 мА. Концентрация каждого фенола в пробе должна быть около 8 мкг [c.469]
Ход определения. Отобранную пробу 25—1000 мл насыщают хлоридом натрия и приливают соляную кислоту до получений 5%-ной концентрации. Затем проводят экстракцию диэтиловым эфиром. Объем эфира и число экстракций зависят от объема взятой для анализа сточной воды. Общим правилом является мне гократное экстрагирование малыми порциями эфира. Все эфир- ные вытяжки соединяют и промывают небольшим объемом раз- бавленной (1 7) соляной кислоты, присоединяя промывные воды к первоначальному раствору. Если на этой стадии анализа получится слишком большой объем эфирного раствора, эфир от-амфотерные органические соединения. [c.255]
Анализ очищенных сточных вод и природных вод, содержащих летучие фенолы в очень малых концентрациях. К дистилляту, полученному из 1 л анализируемой воды, прибавляют 1,5 мл 1 н. раствора едкого натра и насыщают поваренной солью при комнатной температуре. Затем раствор переносят в делительную воронку, добавляют 2 мл 1 н, соляной кислоты и проводят экстракцию, добавляя 50 мл диэтилового эфира и взбалтывая 10 мин. Переносят эфирный слой в небольшую делительную воронку и извлекают из него летучие фенолы, добавляя 10 мл 1,5%-ного раствора едкого кали, и сильно взбалтывают. Весь полученный щелочной раствор используют для получения азокрасителей. Для этого его вносят в маленькую делительную воронку, прибавляют 1 мл разбавленной (1 4) серной кислоты, 10 мл 2 и. раствора карбоната натрия и 1,5 мл раствора диазотированного /г-нитроанилина. После образования смеси кра сителей их извлекают 10 мл разбавленной (1 4) серной кислоты и 5 мл эфира, сильно взбалтывая. [c.381]
Исследование аналогичных производных МДИ и те-траметилендиизоцианата было проведено Ивакурой и Хаяши °. В качестве соединений с активным атомом водорода для получения блокированных изоцианатов ими были использованы фенол, Л4-крезол, диэтиловый эфир [c.139]
Ход определения. К 1 л пробы (Vg) добавляют раствор едкого натра до pH 8—9, раствор переносят в делительную воронку и добавляют 20 мл н-гексана. Воронку сильно встряхивают в течение 5 мин, дают отстояться для разделения слоев и отделяют гек-сановый экстракт. Экстракцию повторяют свежей порцией н-гексана. Вытяжки объединяют и используют для анализов на нефтепродукты и другие органические соединения. К водной части добавляют хлористый натрий (100 г Na l на 1 л пробы) и подкисляют соляной кислотой до pH 3. Полученный раствор в делительной воронке экстрагируют трижды диэтиловым эфиром порциями последовательно по 100, 50 и 50 мл. Эфирные экстракты сливают в одну колбу, сушат безводным сульфатом натрия, переливают в грушевидную колбу и упаривают на водяной бане до объема (Ук) 0,5—1 мл. Из этого объема микрошприцем отбирают пробу (У) от 5 до 20 мкл для анализа в газовом хроматографе. Экстракты фенолов можно хранить в герметично закрытой посуде в течение 12 дней. [c.91]
Введение в молекулу метоксильной и этоксильной групп служит простейшим методом изменения окраски красителя в синий цвет, и поэтому производные анизола, фенетола, толиловых эфиров и диметилового эфира гидрохинона являются важными промежуточными продуктами. Общий метод получения этих эфиров заключается в нагревании щелочного раствора фенола с хлористым алкилом или диалкилсульфатом. Так, анизол (т. кип. 154°) получается с 94% выходом в результате пятичасового нагревания фенола (5 частей), 17,5% раствора едкого натра (15 частей) и хлористого метила (3,6 части) в автоклаве при 125°. Нейтральное масло отделяют, промывают раствором щелочи и очищают вакуум-перегонкой. Диметиловый и диэтиловый эфиры гидрохинона (т. пл. 56° и 71,2°, соответственно) получают из гидрохинона аналогичным способом. Их нитрование 34 % азотной кислотой при 35—85° и восстановление приводит к диметиловому и диэтиловому эфирам 2-амино-гидрохинона, которые служат промежуточными продуктами для Оснований прочно-синих RR и ВВ. [c.146]
ДИФЕНИЛОЛПРОПАН (4,4-диоксидифонилдиме-тилметан, диан) (СПз)2С(СбП40Н)2, мол. в. 228,29 — бесцветные кристаллы т. пл. 156—157° (техпич. продукт 150—152°) т. кип. 250—252/13 м.н растворим в метиловом, этиловом, изопропиловом и бути. го-вом спиртах, в уксусной к-те, ацетоне и в диэтиловом эфире. Растворимость в углеводородах и водо при норма.льной томп-ре незначительная. Получен впервые Дианиным в 1891. В пром-сти Д. получают конденсацией фенола с ацетоном в присутствии серной или соляной к-ты или же хлористого водорода (что дает более чистый продукт) СП СОСН3 - - 2С СНз [c.583]
Крампа [60]. Растворы подкисляли, а азокрасители экстрагировали диэтиловым эфиром до обесцвечивания водного раствора. Полученные экстракты сушили над MgS04 и упаривали эфирно-хлороформенную смесь досуха в струе воздуха. Остаток, содержащий азокрасители, растворяли в хлороформе и доводили до определенного объема. Разделение фенолов, не замещенных по положению 4, проводили методом Крампа, используя в качестве элюента смесь бензола, циклогексана и дипропиленгликоля (30 70 3) и фильтровальную бумагу, пропитанную 20%-ным формами-дом. Для фенолов, замещенных по положению 4, использовали бумагу, импрегнированную смесью ацетона и М,Ы-диметилформ-амида (3 1) [61], и элюент, состоящий из смеси М,Ы-диметил-формамида и гексана (1 4) [62]. [c.192]
В французском патенте, выданном Дрейфусу 14 июля 1911 г. на получение целлулоидонодобных масс из производных целлюлозы, приведены пластификаторы, являющиеся продуктами этерификации одно-или многоатомных фенолов, или нафтолов одноатомными спиртами или фенолами. В частности, в патенте упоминаются анизол, фенетол, простые эфиры крезолов или нафтолов, диэтиловый эфир пирокатехина, причем все эти соединения могут быть замещены в ядре галогеном, нитрогрунпами или теми и другими одновременно. Кроме того, упоминаются простые дифениловый, дибензиловый и метилбензиловый эфиры. Дрейфус пытался пластифицировать и триацетат целлюлозы соединениями этой группы. Среди этих соединений имеется также ряд веществ, которые сейчас квалифицируются как растворители. Они слишком летучи, и это, по-видимому, побудило Дрейфуса предложить в дополнительном патенте применять для той же цели простые эфиры, получаемые из фенолятов и хлорзамещенных этилена или ацетилена. Недавно фирма iba разработала способ получения диэфиров при взаимодействии галогензамещенного метилового эфира алифатических спиртов с фенолами. Например эфир [c.590]
chem21.info
Способ получения диэтилового эфира итаконовой кислоты
„.ЯО„„А
СОЮЗ СОВЕТСКИХ
СОЦИАЛИСТИЧЕ(НИХ
PEGflVSЛИК щ) С 07 С 69 593 р.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Н АВТОРСНОМУ СВИДЕТЕЛЬСТВУ
ГОСУДАРСТВЕННЫЙ КОМИТЕТ СССР
ПО ДЕЛАМ ИЗОБРЕТЕНИЙ И ОТКРЫТИЙ (21) 3259230/23-04 (22) 26,12.80 (46) 30.08.83. Бюл. Р 32 (72) A ° À. Аветисян, Г.Г.Токмаджян, M ° T. Дангян и С.М. Габриелян (71) Ереванский ордена Трудового
Красного Знамени государственный университет (53) 547.391.4 26.07(088.8) (56) 1. Апп. 331, 172, 1904.
2. Патент Фрайции В 1290212, кл. С 07 С, опублик. 1962.
3. J. Am. Cliem. Sos, 70, 3571,1948. (прототип)..
I (54) (57) СПОСОБ ПОЛУЧЕНИЯ ДИЭТИЛОВО-
ГО ЭФИРА ИТАКОНОВОй КИСЛОТЫ, о т л и ч а ю шийся тем, что, с целью упрощения процесса и сокращения его длительности, диэтиловый эфире -этоксаллилянтарной кислоты конденсируют с формальдегидом в присутствии водного раствора поташа при 5-15 С.
1038337
Составитель М. Казанкова
Редактор И. Ковальчук Texpeö A,дч
Корректор й. Тяско г
Заказ 6138/25 Тираж 418 Подписное
ВНИИПИ Государственного комитета СССР по делам изобретений и открытий
113035, Москва, Ж-35, Раушская наб., д.4/5
Филиал ППП "Патент", г. Ужгород, ул. Проектная, 4
Изобретение относится к способу получения диэтилового эфира итаконовой кислоты, который находит применение в производстве термопластиков и сельском хозяйстве, как биостиммуляторы.
Известны способы получения диэтилового эфира,итаконовой кислотй, например взаимодействием итаконовой кислоты с абсолютным этиловым спиртом в присутствии серной кислоты при кипячении (1) .
Однако в,данном случае исходная итаконовая кислота получается из пищевого сырья — сбраживанием сахарозы при помощи Aspergi3.—
1цз 1егг1цз и обработкой культивированной жидкости ионообменными смолами типа сульфокатионитов (2)
Процесс биохимического сбраживания весьма трудоемок и поэтому использование итаконовой кислоты в химических синтезах в значитель"
Ной степени удорожает производство ее различных производных.
Кроме того, известен способ получения диэтилового эфира итаконовой кислоты путем взаимодействия диэтилового эфира фумаровой кислоты с нитрометаном в присутствии диэтиламина (3) .
Однако высокая длительность процесса (11 дней), а также малая доступность самого нитрометана ограничивают возможности этого способа в части .промышленного использования.
Цель изобретения — упрощение процесса и сокращение его длительности .
ПоставЛенная цель достигается тем, что согласно способу получения диэтилового эфира итаконовой ,"кислоты, диэтиловый эфир М -этоксаллилянтарной кислоты конденсируют с формальдегидом в присутствии водного раствора поташа при 5-15
Пример 1. К смеси 21 г (0,076 моль) сырого диэтилового эфира (5-зтоксаллилянтарной кислоты и 10 мл 38%-ного формалина при перемешивании и 15 С прикапывают о раствор 9 г поташа в 50 мл воды.
По окончании прикапывания смесь выдерживают при перемешивании и комнатной температуре 2 ч.
Далее смесь обрабатывают концентрированной соляной кислотой до сильнокислой реакции, экстрагируют эфи10 ром, экстракт высушивают над сульфатом магния и отгоняют эфир, а .остаток перегоняют в вакууме и получают 5,65 г (40%)диэтилового эфира итаконовой кислоты. Т дя. = 7273/2 мм рт. ст., П = 1,4380, а 4 = 1,0450.
Найдено, %: С 58,30, Н 7,.20
С7Н 404
Вычислено, Ъ: С 58,06; Н 7,58
Образец, полученный этерификацией итаконовой кислоты с Т дя 79 С при 3 мм рт.ст. и П > = 1,4380, о идентичен по ГЖХ с полученным в приме ре.
Пример 2. Процесс ведут аналогично примеру 1, но температура прикапывания 5 С. Получают 5,73 г (40,6%) диэтилового эфира итаконовой кислоты с константами, идентичными с ранее полученным.
ЗО Пример 3. Процесс ведут аналогично примеру 1, но температура прикапывания 10 С. Получают
5,63 r (40%) диэтилового эфира итаконовой кислоты с константами, иден,тичными с ранее полученным.
В предлагаемом способе исходный эфир получают конденсацией диэтиловых эфиров янтарной и щавелевой
4П кислот, являющихся промышленно-освоенными продуктами. Способ позволяет также исключить использование пищевого сырья и сократить длительность процесса до 2-х часов, причем процесс ведут в основном при комнатной температуре, что снижает энергетические затраты.
www.findpatent.ru