Окисление спирта, эфира и других веществ перманганатом калия. Окисление простых эфиров перманганатом калия
12. Окисление простых эфиров
Простые эфиры проявляют повышенную склонность к аутоокислению в присутствии кислорода с образованием гидропероксидов. Процесс протекает по цепному радикальному механизму. Эффективным катализатором аутоокисления может служить любой источник свободных радикалов. Аутоокисление простых эфиров представляет большую потенциальную опасность при работе с эфирами в качестве растворителей, поскольку гидропероксиды, накапливающиеся в остатке при перегонке, могут детонировать при слабом перегреве.
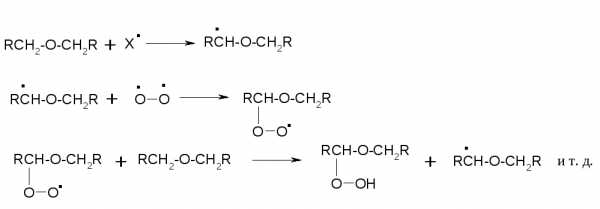
Поэтому гидропероксиды должны быть тщательно удалены до перегонки с помощью восстановителей – солей Fe(II) или Sn(II).
Простые эфиры, имеющие по крайней мере одну первичную алкильную группу, окисляются в соответствующие сложные эфиры с высокими выходами под действием RuO4, а также при действии CrO3 в серной кислоте, перманганата бензилтриэтиламмония и др.:
Простые эфиры енолов окисляются до сложных эфиров под действием хлорхромата пиридиния:
Под действием 1-хлорбензотриазола простые эфиры подвергаются окислительному расщеплению в альдегиды:
13. Окисление эпоксидов
HIO4 расщепляет эпоксиды в альдегиды и кетоны:
При окислении эпоксидов диметилсульфоксидом образуются -гидроксикетоны или -гидроксиальдегиды:
Сильные окислители превращают эпоксиды в кетоны и (или) карбоновые кислоты:
14. Окисление серосодержащих соединений
14.1. Окисление тиолов
Под действием I2, Br2, h3O2, Pb(OAc)4, MnO2 тиолы окисляются до дисульфидов:
2RSH + I2 R-S-S-R
Перкислоты окисляют тиолы до сульфиновых кислот:
Сильные окислители – HNO3 и KMnO4 – окисляют тиолы до сульфоновых кислот:
14.2. Окисление сульфидов
Под действием метапериодата натрия NaIO4, м-хлорнадбензойной кислоты, трет-бутилгипохлорита сульфиды окисляются в сульфоксиды. Наиболее часто применяется 0.5 М водный раствор NaIO4. Этот реагент селективно окисляет сульфиды до сульфоксидов практически без примеси сульфонов, если окисление проводить при 0С в бинарной системе вода – органический растворитель (метанол, диоксан, ацетонитрил):

Механизм окисления аналогичен механизму расщепления 1,2-гликолей и включает циклический интермедиат:
Превращение сульфидов в сульфоксиды под действием трет-бутилгипохлорита можно проиллюстрировать следующим примером:
Окисление сульфидов до сульфонов осуществляется под действием более сильных окислителей (KMnO4, HNO3) или в более жестких условиях при 90-100С с помощью избытка пероксида водорода или трет-бутилгидропероксида в уксусной кислоте:
15. Окисление азотсодержащих соединений
15.1. Окисление аминов
Все амины сравнительно легко окисляются из-за своей основной природы. Легче всего окисляются до N-оксидов третичные амины. В качестве окислителей используют 30%-ный раствор h3O2 в воде, перкислоты в апротонной среде, диоксираны:
При окислении вторичных аминов вслед за образованием N-окиси происходит миграция протона с образованием N,N-диалкилгидроксиламина:
Первичные амины окисляются намного сложнее, поскольку образующееся производное гидроксиламина может далее окисляться до нитрозосоединений:
При наличии атома водорода при -углеродном атоме нитрозосоединение изомеризуется в оксим:
В более жестких условиях первичные амины окисляются до нитросоединений:
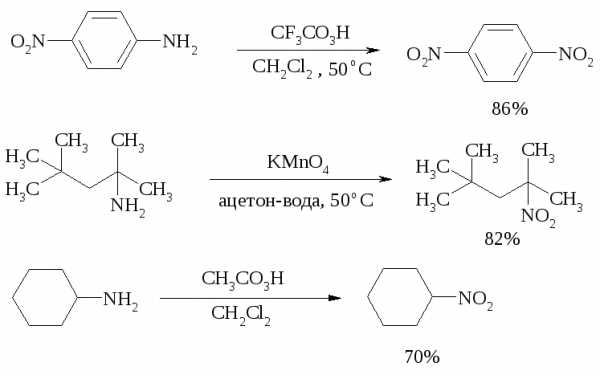
Окисление первичных аминов диметилдиоксираном протекает быстро (от нескольких минут до нескольких часов) в мягких условиях и приводит к соответствующим нитросоединениям с высоким выходом:
Первичные амины, в которых аминогруппа соединена с третичным атомом углерода, с высоким выходом окисляются в нитросоединения перманганатом калия. Первичные амины, содержащие первичные, вторичные или третичные алкильные радикалы окисляются до нитросоединений сухим озоном, а также различными перкислотами.
Первичные, вторичные и третичные алифатические амины расщепляются, давая альдегиды, кетоны или карбоновые кислоты, под действием бромной воды, N-бромсукцинимида, нейтрального раствора KMnO4, нитробензола (для бензиламинов), PdCl2, AuCl3, водного раствора NaOCl в условиях межфазного катализа:
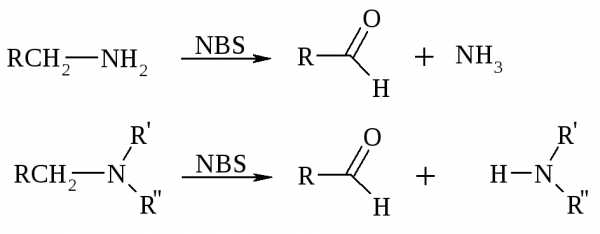
Первичные алифатические амины окисляются в альдегиды или кетоны при взаимодействии с Ag(II), полученным in situ обработкой нитрата серебра персульфатом натрия. Амин сначала дегидрируется до имина, который далее подвергается гидролизу:
Соли бензиламинов дают бензальдегиды или арилкетоны при нагревании в ДМСО:
Дегидрирование первичной аминогруппы, соединенной с первичным атомом углерода, приводит к образованию нитрилов. Реакция осуществлена под действием ряда реагентов: IF5, Pb(OAc)4, CuCl – O2 – пиридин, NBS – (С2H5)3N и др.
Вторичные амины в этих условиях, а также под действием палладиевой черни часто дегидрируются до иминов. Далее имин взаимодействует с молекулой исходного или иного амина, давая аминаль, который теряет аммиак или RNh3, и в результате получается первичный или третичный амин.
Ароматические первичные амины под действием MnO2, Pb(OAc)4, Ba(MnO4)2, O2 в присутствии основания окисляются до азосоединений:
studfiles.net
Окисление эфиров - Справочник химика 21
При окислении эфира образуются также и другие продукты, например уксусный альдегид. Для открытия уксусного альдегида налейте 3—4 капли исследуемого эфира (образец возьмите на общем столе) и добавьте к нему 3 капли раствора фуксинсернистой кислоты (29). При наличии уксусного альдегида постепенно появляется розовое окрашивание, указывающее на разложение эфира. [c.46]
ОКИСЛЕНИЕ ЭФИРА МАРГАНЦЕВЫМ АНГИДРИДОМ [c.129]
На рис. 19 показаны результаты опытов [10] по окислению изопропилового эфира на солнечном свету. Окисление эфира про исходило в стеклянной бутылке на открытом воздухе при температуре 16—28 С. Как видно из графика, концентрация перекиси в эфире растет со временем. [c.72]
Эффективность присадок оценивалась по потере массы масла и изменению его кислотного числа при окислении масляной пленки на поверхности металла. Такие условия в наибольшей мере приближаются к условиям работы иасла в подшипнике, так как при растекании масла по дорожке качения подшипника образуется масляная пленка, которая подвергается воздействию кислорода воздуха при повышенной температуре. Температура испытания была установлена 200°С (на 50 °С выше температуры испытания в подшипнике), продолжительность испытания - 50 ч. В этих условиях окисления эфиры ПЭТ и ЭТР, не содержащие антиокислителей, полностью деструктируются через 50 ч испытания потери массы эфиров достигают 70-80%, оставшаяся на поверхности металлического испарителя часть представляет собой твердую сухую пленку. Введение антиокислителей позволяет в значительной мере затормозить процесс термоокислительной деструкции (табл.1). Как видно, потеря массы афиров, являющаяся следствием двух процессов - физического испарения жидкости и улетучивания легких продуктов термоокислительной деструкции, может быть снижена до — 20%. Оставшаяся в металлическом испарителе часть представляет собой жидкую массу. На стабилизирующий эффект существенно влияет концентрация ингибитора. В данном случае расход ингибитора связан не только с его участием в процессе окисления, но и с его потерей за счет испарения. Возможно поэтому в пределах концентраций от 0,5 до 2% наилучшие результаты были получены при концентрации ингибитора 2%. [c.31]
Путем окисления эфиров сульфиновых кислот в уксуснокислом или бензольном растворе водным раствором перманганата калия или перекисью водорода эфиры сульфокислот получаются с хорошим выходом [c.344]
Лолучение и физические свойства. Взаимодействие ароматических сульфохлоридов с фенолом обычно приводит к арилсульфо-натам. Нормальный ход этой реакции, а также аномальное ее течение с замещением гидроксильной группы на атом галоида подробно описаны в разделе, посвященном сульфо хлоридам (стр. 337). Так как арилсульфонаты исключительно трудно гидролизуются по сравнению с другими эфирами, многие из известных соединений этого типа получены путем нитрования, бромирования и окисления эфиров более простого строения. [c.372]
Эти в высшей степени взрывчатые перекиси образуются в результате самопроизвольного окисления эфира кислородом воздуха (аутоксидация), если эфир хранился некоторое время в соприкосновении с воздухом и на свету. Эфир, который подвергался ранее очистке, но сохранялся затем в течение нескольких месяцев на свету в частично наполненной склянке, т. е. в соприкосновении с воздухом, также может содержать перекиси. [c.59]
ПРИ ОКИСЛЕНИИ ЭФИРОВ до ПЕРЕКИСНЫХ СОЕДИНЕНИИ [c.27]
В табл. 18 приведены алкилсульфонаты, их важнейшие физические свойства и методы получения. В графе Метод получения цифра I означает взаимодействие сульфохлорида со спиртом, II — реакцию серебряной соли сульфокислоты с иодистым алкилом, III — реакцию натриевой соли сульфокислоты с диалкилсульфа-том, IV — окисление эфира сульфиновой кислоты перманганатом калия и V — прочие методы. [c.345]
Реакция Дарзана имеет препаративное значение, так как эфиры эпоксикарбоновых кислот не всегда можно получить окислением эфиров а. -непредельных кислот по Прилежаеву. [c.228]
Окисление эфиров первичных спиртов приводит к альдегидам, карбоновым кислотам, спиртам и в некоторых случаях сложным эфирам [c.258]
Окисленные эфиры жир- Четыреххлористый угле- 2,76 9 [c.10]
Эти данные позволяют сделать интересные выводы о роли различных групп молекулы антиокислителя в процессе окисления эфиров и о механизме самого проц сса окисления эфиров молекулярным кислородом. Однако освещение этого вопроса ие входит в задачу настоящей книги и ниже приводятся лишь заключения [c.257]
В промышленности уже в течение многих лет применяется окисление прямогонных нефтяных остатков, главным образом с целью изменения реологических свойств получаемых из них битумов. В процессе продувки остатков воздухом кислород взаимодействует с компонентами сырья при температуре 200—350 °С. При этом химический состав и соответственно молекулярная структура и свойства остатков изменяются. Соотношение углерод водород для асфальтенов снижается при окислении с 11 1 до 10,5 1. Для смол и масел это соотношение уменьшается, но в меньшей степени (с 8 1 до 7,7 1). Пары воды, двуокись углерода и низкомолекулярные продукты окисления (эфиры, кислоты и альдегиды) удаляются из реакционного объема вместе с продувочными газами. Целевым продуктом является окисленный битум, который существенно отличается от исходного, неокисленного сырья. При окислении изменяется его групповой состав уменьшается содержание масел и значительно возрастает количество асфальтенов, продуктов поликонденсации. Количество силикагелевых смол в некоторых случаях уменьшается, а в других несколько возрастает. [c.32]
Было, однако, замечено, что перевод карбоксильной группы в слох ноэфирную снимает дезактивирующее действие карбоксильной группы. На этой основе разработан четырехстадийный процесс получения диметилтерефталата, состоящий в окислении п-ксилола в гг-толуиловую кислоту, этерификации последней метиловым сиир-гом, окислении эфира п-толуиловой кислоты в моноэфир терефталевой кислоты и его этерификации в диметилтерефталат [c.398]
Эфиры, выходящие с низа эфирнзатора 7, дросселируют и подвергают вакуум-перегонке при остаточном давлении 133 гПа. Вначале в испарителе 8 отгоняют смесь эфиров от менее летучих смолистых примесей. Легкий погон из ректификационной. колонны 10 представляет собой метил-л-толуилат. Он конденсируется в конденсаторе-дефлегматоре 11. Часть его идет на орощение колонны, а остальное количество стекает в сборник 13, откуда направляется на окисление. Эфиры дикарбоновых кислот из куба колонны 10 поступает на вакуум-ректификацию в насадочную колонну 12, где более летучий диметилтерефталат отгоняется от днметиловых эфиров изомерных дикарбоновых кислот ( изофталаты ). В конденсаторе-дефлегматоре 14 эфир конденсируется часть его возвращается на орошение колонны, а остальной продукт стекает в сборник 15. Кубовый остаток из колонны 12 еще содержит значительное количество диметилтерефталата. Его направляют на кристаллизацию из метанольных растворов, на схеме не показанную. Изофталаты лучше растворяются в метаноле, и диметилтерефталат отделяют от них в виде кристаллов, возвращая его на рек-тифика дию. [c.401]
Ж- Дегидрирование янтарной кислоты до фумаровой — единственная реакция, которую нелегко воспроизвести в лаборатории. По типу этот процесс очень похож на ферментативное окисление эфиров жирных кислот с коферментом А при расщеплении жиров (см. выше). Отметим, что в цикле трикарбоновых кислот это дегидрирование стереоспецифично, образуется только транс-изомер и не образуется Г ис-из0мер. [c.261]
Ограниченное использование в синтезе Ф. о. имеют такие методы, как окисление эфиров фосфористой к-ты (с помощью N2O4, Н2О2 и др.), алкилирование солей кислых фосфатов с помощью RHal, алкоголиз или гвдролиз ангидридов фосфорной к-ты, взаимод. алкоксисиланов с галогенвда-ми 4-координац. Р. [c.132]
Окиси эфиров тиоенолов можно легко получить окислением эфиров тиоенолов перекисью водорода. В отличие от сульфидов такие окиси устойчивы к кислотам и снова могут быть восстановлены в эфиры тиоенолов действием алюмогидрида лития. Таким образом можно защитить А -З-кетогруппировку в стероидах во время реакций с участием кислотных реагентов [590]. [c.266]
Раствор переносят в экстрактор и приливают 8—10 мл эфира, содержащего 3,4 М HNO3. Экстрагент готовят встряхиванием эфира с равным объемом 8 М HNO3. Включают мешалку и перемешивают раствор 2—3 мин. Раствор оставляют на 4—5 мин. для полного расслаивания слоев, после чего эфирный экстракт сливают в промывной экстрактор аналогичной конструкции и промывают. Промывным раствором служит 5 М HNO3, насыщенная эфиром. Обычно для этого используют водный слой, оставшийся после приготовления экстрагента. Промытый эфирный экстракт упаривают на водяной бане. Для избежания возможного взрыва в результате окисления эфира концентрированной азотной кислотой добавляют к органическому слою 2—3 мл водного раствора 2 М гидразингидрата, который является более энергичным восстановителем, чем эфир, и поэтому будет предотвращать его окисление. Определение плутония в полученном растворе после отгонки эфира проводят либо сразу радиометрическим методом, либо после проведения дополнительной лантанфторидной очистки. [c.312]
Некоторые хиноны, как например ксилохинон и тимохинон, Бссстакавливаются иодоводородом значительно медленнее, чел бензо-хкнон. В этих случаях реакцию удается довести до конца увеличением избытка иодоводорода. Вместо 2 ж берут б 30%-но1-о раствора иодистого калия и 3 см 30%-нсй серной кислоты. Чтобы предохранить иодоводород от окисления, эфир предварительно насыщают углекислотой II реакцию ведут в атмосфере углекислоты. Эта Л етод ил еет преимущества и при определении бензохинона. [c.345]
Окисление эфира озоном к . В сильно охлажденный абсолютно сухой эфир в присутствии углекислоты пропускается озон. Озонирование сопровождается выделением запаха фруктов. После многочасовой обработки озоном дают в сакуу.ме испариться избыточному эфиру и прн 20 лиг давления перегоняют остаток. При 40 — 50° получается так называемая перекись эфира Бертело в виде бесцветной густой массы, выделяющей при сильном охлаждении из приемника небольшое количество бесцветного кристаллического вещества. При попадании воздуха в эвакуированный пере1 онный сосуд может иногда произойти сильный Езрью вообще получаемое прн этом вещество отличается необычайно сильными взрывчатыми свойсткамн. [c.104]
Из ЭТИХ данных татгже следует, что атом углерода в у-положе-нии уже не испытывает практически никакого защитного дей СТВ1Ш карбонильной группы. Кроме того, можно сделать дополнительно вывод о том, что скорость окисления эфиров адипиновой кислоты лишь незначительно увеличивается ( в 1,5 раза) при увеличении длины спиртового радикала в 3 раза. [c.130]
Стойкость к окислению эфиров глутаровой, адипиновой, соба-циновой кислот и 1/ -сииртов значительно увеличивается ири добавлении 0,5% фентиазина ири 163° и 1,5% — ири 200°, хотя при 200° наблюдаются отложевия на металлах. Ди-(1/ -амил) глутарат ири 200° с добавкой 0,2% фентиазина весьма стоек к окислению, осадок ири окислении его был очень незначителен. [c.199]
Антиокислители для сложных эфиров карбоновых кислот [26, 33, 34, 35]. Подбор антиокислителей для сложных эфиров карбоновых кислот и как предпосылка к этому изучение услог.ий окисления эфиров исследовались, как указывалось ранее, достаточно широко. Изучались многие представители группы фенолов, аминов, тиодифениламина, соединений, содержащих серу, фосфор, металлы и некоторые другие элементы. Описываются испытания более 100 соединений. [c.253]
Добавка фентиазина ( 0,5% вес.) при температуре до 150— 163° задерживает приблизительно в одинаковой мере окисление эфиров двухосновных кислот (эфиров трикарбаллиловой кислоты и ряда эфиров многоатомных спиртов). Окисление эфиров аконитовой кислоты уже при 125° не задернсивается применением фентиазина. [c.255]
Окисление эфиров 4 ннтробензолсульфокнслоты [5]. Окисление первичных спиртов до альдегидов с помощью ДМСО требует температур около 100° или добавления кислот или ионов тяжелых металлов. По новой методике спирт действием 4-нит-робензолсульфохлорида и пиридина сначала превращают в эфир 4-нитробензолсульфокислоты, а затем полученный эфир окисляют ДМСО в присутствии бикарбоната натрия. Реакцию проводят при комнатной температуре при перемешивании в течение около 100—200 час. [c.161]
chem21.info
Окисление спирта, эфира и других веществ перманганатом калия
а) На жестяную крышку, кусок жести или крышку от фарфорового тигля с отбитым ушком, помещенную на асбестированную сетку, насыпать около 1 гмелких кристалликов перманганата калия и осторожно облить из капельной («глазной») пипетки концентрированной серной кислотой так, чтобы масса не растекалась. Из пипетки приливать к смеси по каплям спирт (лучше с небольшой добавкой серного эфира). Каждая капля вызывает появление пламени.
б) На приготовленную изложенным выше способом окислительную смесь выжать спирт из комка ваты. Для этого обильно смоченную спиртом вату зажать в руке и как бы взмахнуть ею над окислительной смесью (на высоте не менее 50 см), нажать при этом пальцами на вату, чтобы выжимаемый спирт попал на смесь. Появляется большое пламя, которое после сгорания спирта гаснет само собой. Поблизости не должно быть легковоспламеняющихся материалов.
в) Можно взять некоторое количество окислительной смеси на стеклянную палочку и коснуться ею бумаги или ваты, смоченных смесью спирта с эфиром.
Вместо спирта можно взять бензин.
г) Фитиль стеклянной спиртовки обильно смочить спиртом и насыпать на него измельченный перманганат калия. Конец стеклянной палочки обмакнуть на 0,5 смв концентрированную серную кислоту и прикоснуться им к фитилю; спиртовка загорается.
д) Пламя из стеклянной трубки. Для опыта нужна стеклянная трубка длиной 35 см, диаметром 1 см, согнутая на 13 длины под углом около 20°. В трубку до места сгиба плотно вставляют просвер-
ленную (отверстие диаметром около 3 мм) корковую пробку .
Перед демонстрацией в чистый фарфоровый тигель, помещенный (обязательно!) Б стеклянную пол-литровую банку, насыпают около половины чайной ложки измельченного перманганата калия, осторожно пипеткой наливают столько серной кислоты, чтобы получилась жидкая кашица (неперемешивать!),и закрывают банку стеклянной пластинкой, предо.храняющей от вылетания брызг и хлопьев твердых продуктов реакции в случае самопроизвольного разложения. Рыхлый комок ваты обильно смачивают серным эфиром и кладут на стол (можно прикрыть стаканом). Сдвинув стекло с банки, опускают короткий (от места сгиба) конец трубки в окислитать-ную смесь и слегка поворачивают трубку, без нажима на дно тигля. На конце трубки остается после этого зеленое или темноватое кольцо шириной около 0,5 см. Вынув трубку и закрыв банку, следует возможно быстрее взять смоченную эфиром вату, вставить ее в длинный конец трубки, протолкнуть стеклянной палочкой почти до пробки и, взяв этот конец трубки в рот, сильно подуть в него. У короткого конца трубки появляется большое пламя. Вдувание воздуха с небольшими перерывами можно повторить несколько раз. Трубку после опыта хорошо вымыть (особенно короткий конец) и просушить (пробку можно не вынимать). Тигель с оставшейся в нем окислительной смесью залить водой, не вынимая из банки. Повторять опыт в случае неудачи и т. п. с невымытон или мокрой трубкой нельзя. Не рекомендуется демонстрировать опыт, не вставив в трубку просверленную пробку: при вдувании воздуха вата может вылететь из трубки и воспламениться. Посуда, реактивы должны быть чистыми, попадание случайных, особенно горючих, примесей может вызвать преждевременное разложение окислительной смеси и даже взрыв.
Более просто можно нанести окислитель на конец трубки, погрузив ее на 1 в тигель (стаканчик) с концентрированной серной кислотой, а затем, держа трубку вертикально, прижать смоченный конец ее к мелкому порошку перманганата калия, насыпанному в другой тигель слоем не менее 0,5 см. Но при таком нанесении окислителя опыт ие удается значительно чаще, чем в первом варианте.
class.in.ua
Окислительно-восстановительные реакции с участием органических веществ » HimEge.ru
В окислительно-восстановительных реакциях органические вещества чаще проявляют свойства восстановителей, а сами окисляются. Легкость окисления органических соединений зависит от доступности электронов при взаимодействии с окислителем. Все известные факторы, вызывающие увеличение электронной плотности в молекулах органических соединений (например, положительные индуктивный и мезомерные эффекты), будут повышать их способность к окислению и наоборот.
Склонность органических соединений к окислению возрастает с ростом их нуклеофильности, что соответствует следующим рядам:
Рост нуклеофильности в ряду
Рассмотрим окислительно-восстановительные реакции представителей важнейших классов органических веществ с некоторыми неорганическими окислителями.
Окисление алкенов
При мягком окислении алкены превращаются в гликоли (двухатомные спирты). Атомы-восстановители в этих реакциях – атомы углерода, связанные двойной связью.
Реакция с раствором перманганата калия протекает в нейтральной или слабо щелочной среде следующим образом:
3C2h5 + 2KMnO4 + 4h3O → 3Ch3OH–Ch3OH + 2MnO2 + 2KOH
В более жестких условиях окисление приводит к разрыву углеродной цепи по двойной связи и образованию двух кислот (в сильно щелочной среде – двух солей) или кислоты и диоксида углерода (в сильно щелочной среде – соли и карбоната):
1) 5Ch4CH=CHCh3Ch4 + 8KMnO4 + 12h3SO4 → 5Ch4COOH + 5C2H5COOH + 8MnSO4 + 4K2SO4 + 17h3O
2) 5Ch4CH=Ch3 + 10KMnO4 + 15h3SO4 → 5Ch4COOH + 5CO2 + 10MnSO4 + 5K2SO4 + 20h3O
3) Ch4CH=CHCh3Ch4 + 8KMnO4 + 10KOH → Ch4COOK + C2H5COOK + 6h3O + 8K2MnO4
4) Ch4CH=Ch3 + 10KMnO4 + 13KOH → Ch4COOK + K2CO3 + 8h3O + 10K2MnO4
Дихромат калия в сернокислотной среде окисляет алкены аналогично реакциям 1 и 2.
При окислении алкенов, в которых атомы углерода при двойной связи содержат по два углеродных радикала, происходит образование двух кетонов:
Окисление алкинов
Алкины окисляются в несколько более жестких условиях, чем алкены, поэтому они обычно окисляются с разрывом углеродной цепи по тройной связи. Как и в случае алкенов, атомы-восстановители здесь – атомы углерода, связанные кратной связью. В результате реакций образуются кислоты и диоксид углерода. Окисление может быть проведено перманганатом или дихроматом калия в кислотной среде, например:
5Ch4C≡CH + 8KMnO4 + 12h3SO4 → 5Ch4COOH + 5CO2 + 8MnSO4 + 4K2SO4 + 12h3O
Ацетилен может быть окислен перманганатом калия в нейтральной среде до оксалата калия:
3CH≡CH +8KMnO4→ 3KOOC –COOK +8MnO2 +2КОН +2Н2О
В кислотной среде окисление идет до щавелевой кислоты или углекислого газа:
5CH≡CH +8KMnO4 +12h3SO4 → 5HOOC –COOH +8MnSO4 +4К2SO4 +12Н2ОCH≡CH + 2KMnO4 +3h3SO4 → 2CO2 + 2MnSO4 + 4h3O + K2SO4
Окисление гомологов бензола
Бензол не окисляется даже в довольно жестких условиях. Гомологи бензола могут быть окислены раствором перманганата калия в нейтральной среде до бензоата калия:
C6H5Ch4 +2KMnO4 → C6H5COOK + 2MnO2 + KOH + h3O
C6H5Ch3Ch4 + 4KMnO4 → C6H5COOK + K2CO3 + 2h3O + 4MnO2 + KOH
Окисление гомологов бензола дихроматом или перманганатом калия в кислотной среде приводит к образованию бензойной кислоты.
5С6Н5СН3+6КMnO4+9 h3SO4→ 5С6Н5СООН+6MnSO4 +3K2SO4 + 14h3O
5C6H5–C2H5 + 12KMnO4 + 18h3SO4 → 5C6H5COOH + 5CO2 + 12MnSO4 + 6K2SO4 + 28h3O
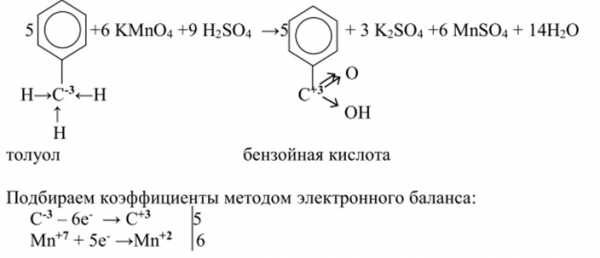
Окисление спиртов
Непосредственным продуктом окисления первичных спиртов являются альдегиды, а вторичных – кетоны.
Образующиеся при окислении спиртов альдегиды легко окисляются до кислот, поэтому альдегиды из первичных спиртов получают окислением дихроматом калия в кислотной среде при температуре кипения альдегида. Испаряясь, альдегиды не успевают окислиться.
3C2H5OH + K2Cr2O7 + 4h3SO4 → 3Ch4CHO + K2SO4 + Cr2(SO4)3 + 7h3O
С избытком окислителя (KMnO4, K2Cr2O7) в любой среде первичные спирты окисляются до карбоновых кислот или их солей, а вторичные – до кетонов.
5C2H5OH + 4KMnO4 + 6h3SO4 → 5Ch4COOH + 4MnSO4 + 2K2SO4 + 11h3O
3Ch4–Ch3OH + 2K2Cr2O7 + 8h3SO4 → 3Ch4–COOH + 2K2SO4 + 2Cr2(SO4)3 + 11h3O
Третичные спирты в этих условиях не окисляются, а метиловый спирт окисляется до углекислого газа.
Двухатомный спирт, этиленгликоль HOCh3–Ch3OH, при нагревании в кислой среде с раствором KMnO4 или K2Cr2O7 легко окисляется до щавелевой кислоты, а в нейтральной – до оксалата калия.
5СН2(ОН) – СН2(ОН) + 8КMnO4+12h3SO4→ 5HOOC –COOH +8MnSO4 +4К2SO4 +22Н2О
3СН2(ОН) – СН2(ОН) + 8КMnO4→ 3KOOC –COOK +8MnO2 +2КОН +8Н2О
Окисление альдегидов и кетонов
Альдегиды – довольно сильные восстановители, и поэтому легко окисляются различными окислителями, например: KMnO4, K2Cr2O7, [Ag(Nh4)2]OH, Cu(OH)2. Все реакции идут при нагревании:
3Ch4CHO + 2KMnO4 → Ch4COOH + 2Ch4COOK + 2MnO2 + h3O
3Ch4CHO + K2Cr2O7 + 4h3SO4 → 3Ch4COOH + Cr2(SO4)3 + 7h3O
Ch4CHO + 2KMnO4 + 3KOH → Ch4COOK + 2K2MnO4 + 2h3O
5Ch4CHO + 2KMnO4 + 3h3SO4 → 5Ch4COOH + 2MnSO4 + K2SO4 + 3h3O
Ch4CHO + Br2 + 3NaOH → Ch4COONa + 2NaBr + 2h3O

реакция «серебряного зеркала»
C аммиачным раствором оксида серебра альдегиды окисляются до карбоновых кислот которые в аммиачном растворе дают соли аммония (реакция «серебрянного зеркала»):
Ch4CH=O + 2[Ag(Nh4)2]OH → Ch4COONh5 + 2Ag + h3O + 3Nh4
Ch4–CH=O + 2Cu(OH)2 → Ch4COOH + Cu2O + 2h3O
Муравьиный альдегид (формальдегид) окисляется, как правило, до углекислого газа:
5HCOH + 4KMnO4(изб) + 6h3SO4 → 4MnSO4 + 2K2SO4 + 5CO2 + 11h3O
3СН2О + 2K2Cr2O7 + 8h3SO4 → 3CO2 +2K2SO4 + 2Cr2(SO4)3 + 11h3O
HCHO + 4[Ag(Nh4)2]OH → (Nh5)2CO3 + 4Ag↓ + 2h3O + 6Nh4
HCOH + 4Cu(OH)2 → CO2 + 2Cu2O↓+ 5h3O
Кетоны окисляются в жестких условия сильными окислителями с разрывом связей С-С и дают смеси кислот:
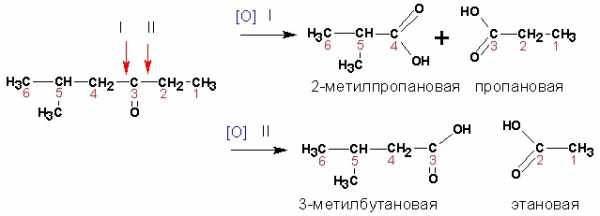
Карбоновые кислоты. Среди кислот сильными восстановительными свойствами обладают муравьиная и щавелевая, которые окисляются до углекислого газа.
НСООН + HgCl2 =CO2 + Hg + 2HCl
HCOOH+ Cl2 = CO2 +2HCl
HOOC-COOH+ Cl2 =2CO2 +2HCl
Муравьиная кислота, кроме кислотных свойств, проявляет также некоторые свойства альдегидов, в частности, восстановительные. При этом она окисляется до углекислого газа. Например:
2KMnO4 + 5HCOOH + 3h3SO4 → K2SO4 + 2MnSO4 + 5CO2↑ + 8h3O
При нагревании с сильными водоотнимающими средствами (h3SO4 (конц.) или P4O10) разлагается:
HCOOH →(t) CO↑ + h3O
Каталитическое окисление алканов:
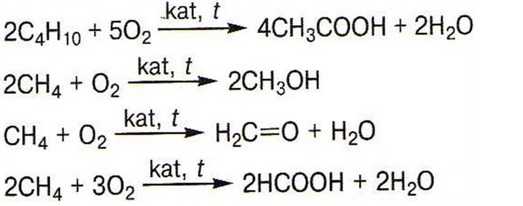
Каталитическое окисление алкенов:
Окисление фенолов:
himege.ru
3. Окисление алкенов
3.1. Синтез эпоксидов
Под действием перкислот (перуксусной, пермуравьиной, м-хлорпербензойной, пертрифторуксусной и др.) алкены окисляются в α-окиси, или оксираны (реакция Прилежаева). Электронодонорные заместители при двойной связи облегчают протекание реакции. Процесс окисления протекает стереоспецифично, и α-окись сохраняет ту же конфигурацию, что и исходный алкен.
Процесс носит синхронный, согласованный характер и протекает по схеме:
Альтернативный метод эпоксидирования заключается во взаимодействии алкена с нитрилом и 90%-ным пероксидом водорода:
Окись этилена получают прямым окислением этилена кислородом воздуха в присутствии серебра, нанесенного на оксид алюминия или карбид кремния:
Окись этилена легко отделяется от этилена при растворении в воде с последующей перегонкой.
Все попытки получения окиси пропилена прямым окислением пропилена кислородом на серебряном катализаторе были безуспешны, поскольку окислению подвергались С-Н-связи метильной группы в аллильном положении к двойной связи. В результате был разработан альтернативный промышленный метод синтеза окиси пропилена, известный под названием халкон-процесса. В халкон-процессе пропилен окисляется до его оксида под действием трет-бутилгидропероксида. Необходимый гидропероксид образуется в результате окисления изобутана в жидкой фазе кислородом при 120-150ºС и давлении 3 МПа. Далее он реагирует с пропиленом в жидкой фазе при 120-140ºС и давлении 3.5 МПа в присутствии солей молибдена как катализатора.
Выход окиси пропилена в расчете на пропилен составляет 90%.
Эпоксиды могут быть получены с высоким выходом при действии на алкены диоксиранов:
3.2. Вакер-процесс
В Вакер-процессе этилен окисляют в водном растворе хлористоводородной кислоты, содержащем хлориды палладия (II) и меди (II). Протекающие при этом реакции описываются следующими уравнениями:
или суммарно:
Таким образом, в процессе окисления расходуется только кислород.
Помимо этилена многие другие монозамещенные и 1,2-дизамещенные алкены окисляются в альдегиды и кетоны хлоридом палладия. В случае 1,1-дизамещенных алкенов обычно получаются неудовлетворительные результаты.
Если вместо воды в Вакер-процессе использовать уксусную кислоту, то образуется винилацетат:
Смесь этилена и уксусной кислоты окисляется в газовой фазе в присутствии палладиевого катализатора при 200ºС и давлении 10 атм, выход винилацетата достигает 90-95%.
3.3. Синтез вицинальных диолов
При взаимодействии алкенов в слабощелочной среде с разбавленным раствором KMnO4 в водных органических растворителях (ацетоне, этаноле и др.) при 0-10ºС образуются вицинальные диолы с выходами 30-75% (реакция Вагнера).
Первоначально образуется циклический эфир марганцевой кислоты, который немедленно гидролизуется до диола:
Эффективным гидроксилирующим агентом является тетраоксид осмия OsO4. При действии на алкены эквимолярного количества OsO4 в абсолютном эфире, пиридине или бензоле при комнатной температуре медленно образуется продукт присоединения, который при гидролизе водным раствором NaHSO3 или сероводородом дает с высокими выходами 1,2-диолы.
Тетраоксид осмия может использоваться и как катализатор в реакции окисления. Это позволяет работать лишь с незначительными его количествами, что важно вследствие высокой токсичности и стоимости данного реагента. Окисление проводят 30%-ным водным раствором пероксида водорода в трет-бутаноле при 0ºС.
Все гликоли, полученные окислением OsO4 или KMnO4, имеют цис-конфигурацию. Следует отметить, что высшие оксиды других переходных металлов (V2O5, WO3, MoO3 и др.) катализируют анти-гидроксилирование алкенов. Продукты антигидроксилирования алкенов можно также получить гидролизом соответствующих эпоксидов, либо в одном процессе совместить стадии образования и расщепления эпоксида, если алкен обрабатывать водным 30-70%-ным пероксидом водорода в муравьиной или трифторуксусной кислоте:
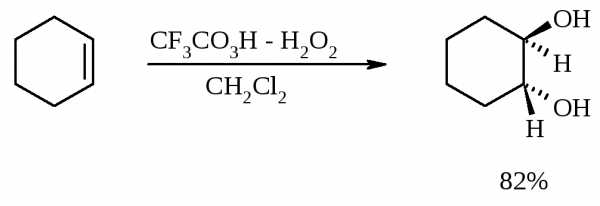
Кроме того, продукты транс-присоединения к двойной связи можно получить при обработке алкена иодом и бензоатом или ацетатом серебра в безводном бензоле или CCl4(реакция Прево). Первоначально происходит образование йодэфира, в котором иод далее замещается бензоат-ионом.
Последующий гидролиз дибензоата гликоля приводит к 1,2-диолу.
studfiles.net
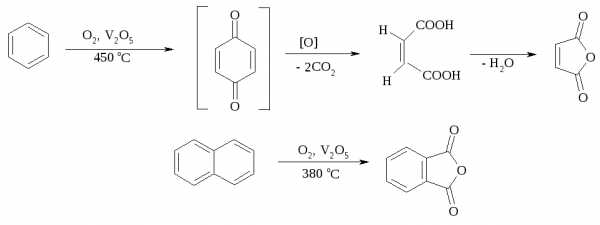
5.3. Окислительное расщепление аренов
В промышленности окислением бензола и нафталина кислородом воздуха в присутствии V2O5 получают соответственно малеиновый и фталевый ангидриды:
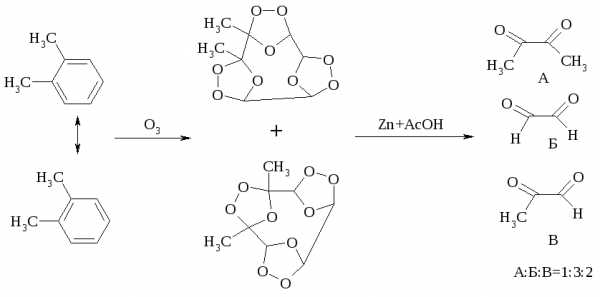
При взаимодействии бензола и алкилбензолов с озоном образуются триозониды, которые далее подвергают окислительному или воcстановительному расщеплению:
Препаративное значение имеет также окисление нафталина KMnO4, приводящее к о-формилбензойной кислоте.
В замещенных нафталинах действие окислителя направляется на кольцо с большей электронной плотностью.

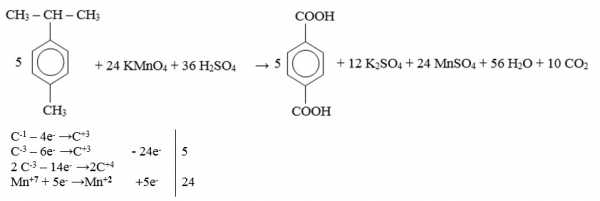
5.4. Окисление боковых цепей в ароматических соединениях
Окисление алкиларенов происходит в первую очередь по бензильному атому углерода, что объясняется легкостью образования соответствующего бензильного радикала.
Для окисления боковых цепей в алкилбензолах в карбоксильную группу применяют водный раствор KMnO4 при нагревании, смесь Na2Cr2O7 и h3SO4.

Следует особо отметить, что в этих условиях любая алкильная группа, содержащая атомы водорода в положении по отношению к бензольному кольцу, окисляется до карбоксильной.
Если алкильная группа не содержит атомов водорода в положении по отношению к бензольному кольцу, такая трет-алкильная боковая группа не окисляется под действием Na2Cr2O7или KMnO4 в кислой или нейтральной среде. Так, например, трет-бутилбензол окисляется в очень жестких условиях перманганатом калия до триметилуксусной кислоты, т. е. окисляется само бензольное кольцо:
Однако водная азотная кислота окисляет трет-алкильные группы до карбоксильных.
Дихромат натрия и перманганат калия нерастворимы в ароматических углеводородах, поэтому окисление идет в гетерогенных условиях, что часто резко снижает выход продуктов окисления. Этого недостатка лишен метод межфазного переноса реагентов. Твердый перманганат калия частично растворяется в бензоле в присутствии 18-краун-6-полиэфира вплоть до концентрации 0.06 М. Такой раствор носит название "пурпурный (малиновый) бензол" и широко используется для окисления алкилбензолов:
При использовании же в качестве катализатора межфазного переноса бромида тетрабутиламмония практически весь (95%) перманганат-ион находится в органической фазе.
При синтезе аминобензойных кислот из алкиланилинов, содержащих первичную аминогруппу, используют ацильную защиту:
Алкильную группу в алкилнафталинах можно окислить до карбоксильной, не затронув при этом нафталиновые ядра, если использовать в качестве окислителя нейтральный водный раствор дихромата натрия и проводить реакцию при высоких температурах в автоклаве:
Для окисления метилбензолов в альдегиды используют смесь хромового и уксусного ангидридов. При этом образующийся альдегид превращается в ацилаль, который далее не окисляется. Омылением этого производного серной кислотой получают сам альдегид. Орто-замещенные бензальдегиды получаются таким путем с низкими выходами.
Другой способ превращения метилзамещенных бензолов в бензальдегиды состоит в их обработке хлористым хромилом в CCl4 или CS2 (реакция Этара). Вначале окисляемое вещество образует комплекс с двумя молекулами CrO2Cl2, который выпадает в осадок. При обработке комплекса водой образуется альдегид.
Данный метод дает возможность окислять только одну метильную группу в присутствии других:
Окисление метиленового звена в алкилбензолах с образованием кетонов можно провести под действием ряда окислителей (разб. HNO3, SeO2, Na2Cr2O7 и др.). Особенно легко окисляются диарилметаны:
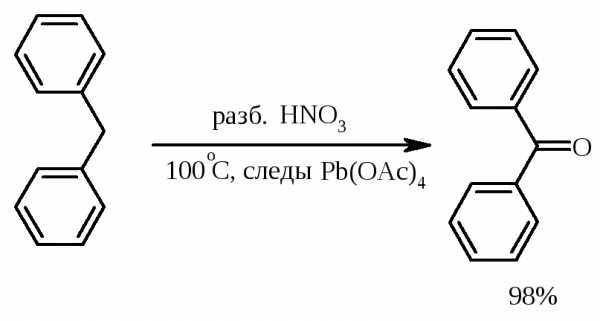
Окисление жирноароматических соединений можно осуществлять таким образом, чтобы окислению кислородом воздуха подвергался карбанион, который образуется при депротонировании исходной СН-кислоты в инертной апротонной среде (ТГФ, диметоксиэтане). Жирноароматических соединений с pKaниже 33-35 можно окислить в системе КОН - 18-краун-6 - ТГФ до ароматических кислот, кетонов и триарилкарбинолов:
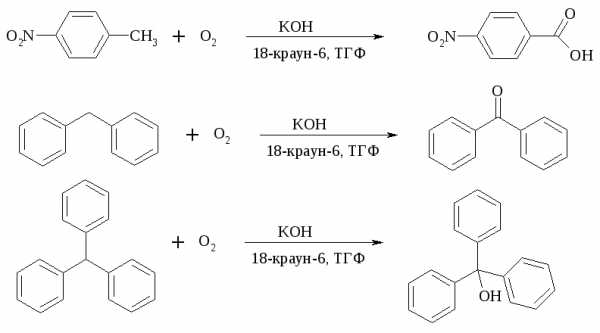
Окисление кумола в гидропероксид с последующим разложением его серной кислотой приводит к образованию ацетона и фенола:
Аналогично из индана и тетралина можно получить соответствующие кетоны:
Важнейшее промышленное значение имеют реакции прямого окисления о- и n-ксилолов кислородом воздуха до фталевой и терефталевой кислот соответственно в присутствии ацетата кобальта (III) в уксуснокислом растворе:
Введение ацетоксигруппы в бензильное положение алкилбензолов обычно осуществляют с помощью тетраацетата свинца. Реакцию проводят кипячением смеси реагентов в бензоле или ледяной уксусной кислоте. Процесс протекает по свободнорадикальному механизму:
Соответствующие спирты получают последующим омылением ацетатов.
studfiles.net
11. Окисление карбоновых кислот
При действии пероксида водорода в присутствии кислотного катализатора на карбоновые кислоты образуются перкислоты (надкислоты):
Наиболее распространенным катализатором в случае алифатических карбоновых кислот является концентрированная h3SO4. Реакция обратима, и равновесие можно сместить вправо, удаляя воду или применяя избыток реагента. Для субстратов с ароматическими группами R наилучшим катализатором является метансульфокислота, которая используется и как растворитель.
Карбоновые кислоты подвергаются окислительному декарбоксилированию под действием тетраацетата свинца и в качестве продуктов в зависимости от условий получаются алканы, алкены или эфиры уксусной кислоты:
Для данной реакции предполагается следующий механизм:
Алканы образуются за счет отрыва атома водорода от молекул растворителя радикалом R, а алкен и сложный эфир из карбокатиона соответственно за счет отщепления протона или захвата ацетат-иона. Введение в реакционную смесь галогенид-иона практически нацело подавляет оба процесса и приводит к образованию алкилгалогенидов.
В присутствии иода и тетраацетата свинца карбоновые кислоты превращаются в соответствующие иодиды:
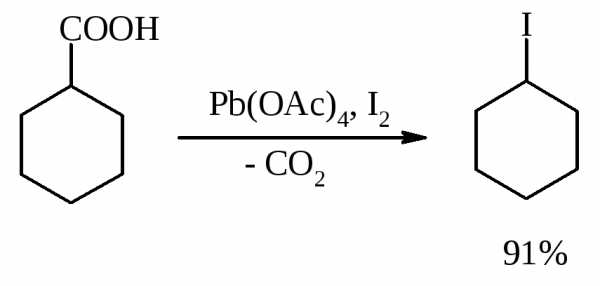
Из 1,2-дикарбоновых кислот под действием тетраацетата свинца образуются алкены:
Другим примером окислительного декарбоксилирования может служить окисление серебряных солей карбоновых кислот бромом в CCl4 с образованием алкилбромидов (реакция Хунсдиккера – Бородина):
Для успешного проведения реакции требуется применять тщательно высушенные серебряные соли карбоновых кислот, и выход алкилбромида колеблется в широких пределах в зависимости от степени очистки и обезвоживания соли. Этого недостатка лишена модификация с использованием ртутных солей, причем ртутную соль не выделяют индивидуально, а смесь карбоновой кислоты, оксида ртути (II) и брома нагревают в индифферентном растворителе. Этот метод приводит, как правило, к более высоким и воспроизводимым выходам.
Для реакции Хунсдиккера – Бородина установлен цепной радикальный механизм. Образующийся в первой стадии ацилгипобромит подвергается гомолитическому расщеплению с образованием карбоксильного радикала и атома брома. Карбоксильный радикал теряет CO2 и превращается в алкильный радикал, который затем регенерирует цепь, отщепляя атом брома от ацилгипобромита.

-Гидрокси- и -кетокислоты не расщепляются под действием HIO4, но эта реакция идет с тетраацетатом свинца, h3O2 в щелочной среде и другими реагентами. Такие реакции представляют собой окислительное декарбоксилирование. Из -гидроксикислот получаются альдегиды или кетоны, а из -кетокислот – карбоновые кислоты:
В то же время при использовании пероксида водорода в присутствии Fe(III) удается окислить гидроксильную группу в -гидроксикислотах в карбонильную с сохранением карбоксильной:
Окислить алифатические карбоновые кислоты в α-гидроксикислоты удается в том случае, если кислота содержит третичный атом углерода в α-положении к карбоксильной группе. В качестве окислителя применяют щелочной раствор KMnO4.
studfiles.net